- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of in vivo Cellulose Biosynthesis in Arabidopsis Cells by Spinning Disk Confocal Microscopy
(*contributed equally to this work) Published: Vol 5, Iss 19, Oct 5, 2015 DOI: 10.21769/BioProtoc.1617 Views: 9510
Reviewed by: Fanglian HeRenate Weizbauer
Original research article
The authors used this protocol in:

Jun 2006
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Cellulose is a main component of plant cell walls. Tools to analyze cellulose mainly rely on analytical chemistry, which yields information about cellulose amounts and structure, but cannot be applied to intact tissues. Moreover, these methods measure total cellulose and cannot be used to assay cellulose synthesis per se. Live cell imaging of the catalytic subunits of the cellulose synthesis complex (CSC) conjugated to fluorescent proteins is an important tool to understand the dynamics of the cellulose biosynthesis process (Paredez et al., 2006). This method can be used in various genetic backgrounds (Sorek et al., 2014) or with different chemical inhibitors (Brabham and Debolt, 2012). Here we describe in detail the procedure to visualize the movement of CSCs at the plasma membrane. As the movement of CSCs is likely caused by glucan synthesis and extrusion into the cell wall, live cell analysis of CSC velocity provides a method to directly measure cellulose synthesis in vivo.
Keywords: CelluloseMaterials and Reagents
- Microscope slides (25 x 76 x 1.0 mm) and #1.5 cover glass (24 x 30 mm)
- Arabidopsis seedlings expressing functional fluorescent protein fusions to CESAs, the catalytic subunits of the CSC, such as GFP:CESA3 (Desprez et al., 2007), YFP:CESA6 (Paredez et al., 2006) or tdTomato:CESA6 (Sánchez-Rodríguez et al., 2012) under the control of their native promoters
- Household bleach (Clorox)
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 71727 )
- Murashige and Skoog (MS) basal salts (Caisson Laboratories, catalog number: MSP01 )
- 2-(N-morpholino)ethanesulfonic acid (MES) (Sigma-Aldrich, catalog number: RES0113M-B103X )
- Sucrose (Fisher Scientific, catalog number: BP220 )
- Agar (Sigma-Aldrich, catalog number: RES10020-A102X )
- Vacuum Grease (Beckman Coulter)
- 0.5x Murashige and Skoog (MS) media (see Recipes)
Equipment
- Growth chamber to grow plant material (e.g., Percival Scientific, model: CU-36L5 )
- Square plates 90 x 90 x 15 mm
- Spinning disk confocal head (Yokogawa Electric Corporation) mounted on a motorized inverted microscope (e.g., Leica Microsystems, model: Leica DMI6000 or Zeiss, model: Zeiss Cell Observer SD ), equipped with 488 and/or 561 nm excitation lasers and a Photometrics QuantEM 512SC Camera
Software
- Software operating the confocal microscope (e.g., Metamorph, Molecular Devices)
- ImageJ (http://imagej.nih.gov/ij/)
- MultipleKymograph and WalkingAverage plugins for ImageJ (J. Reitdorf and A. Seitz, http://www.embl.de/eamnet/html/body_kymograph.html)
- Imaris (BitPlane)
- Excel (Microsoft)
Procedure
- Plant growth
- Sterilize Arabidopsis seeds in 30% (v/v) household bleach (final concentration ~1.5% w/v sodium hypochlorite) and 0.1% (w/v) SDS for 15 min and wash three times with sterile water.
- Stratify the seeds in sterile 0.15% (w/v) agar at 4 °C for at least three days in the dark and sow on 0.5x MS, 0.8% agar, 1% sucrose plates.
- After two hours of light exposure at room temperature, wrap the plates in aluminum foil and grow the seedlings vertically in the dark for 3 days at 22 °C.
- Sterilize Arabidopsis seeds in 30% (v/v) household bleach (final concentration ~1.5% w/v sodium hypochlorite) and 0.1% (w/v) SDS for 15 min and wash three times with sterile water.
- Sample preparation (Figure 1)
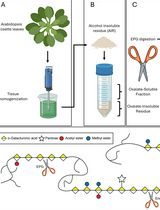
- Apply a thin layer of vacuum grease (in syringe) at four corners near the edges of a 25 x 75 x 1 mm slide (Figure 1A) to create a mounting chamber. The vacuum grease cushion stabilizes the seedling for sustained time-lapse imaging, minimizing compression and mechanical damage.
- Transfer an Arabidopsis etiolated seedling into the slide with 135 μl of water or liquid 0.5x MS media (Figure 1B) and mount with a #1.5 24 x 30 mm cover glass (Figure 1C). Manipulate the seedling as quickly as possible, or use a safer light such as green or red light, to minimize exposure to light.
- Apply a thin layer of vacuum grease (in syringe) at four corners near the edges of a 25 x 75 x 1 mm slide (Figure 1A) to create a mounting chamber. The vacuum grease cushion stabilizes the seedling for sustained time-lapse imaging, minimizing compression and mechanical damage.
- Confocal imaging
- Observe the seedling under a confocal microscope featuring a spinning disk head mounted on a motorized inverted microscope with a 100x/1.4 numerical aperture oil immersion objective.
- Use 488 nm or 561 nm excitation wavelength for the fluorescent markers GFP/YFP and tdTomato respectively; and band pass filters (525/50 nm for GFP, 535/50 for YFP, and 620/60 for tdTomato) for emission filtering. Perform imaging with 500-ms exposure times.
- Obtain CESA particle images from epidermal cells in the upper etiolated hypocotyl (Figure 2A) at the plasma membrane focal plane (Figure 2B), where cellulose synthesis takes place. The larger particles in the adjacent focal planes correspond to CESA particles in Golgi and cytoplasmic compartments (Figure 2C).
- Run a time-lapse imaging experiment with a time interval of 5 sec and duration of 5 min.
- During this time, vertical stage drift may occur, resulting in a shift from the plasma membrane focal plane. Make sure that images are acquired at the correct focal plane over time. This can be accomplished using a “live” preview mode every four or five frames to adjust the focus. Since this can increase photo bleaching of the sample, adjustments should be made only when necessary.
- To check that there is no horizontal stage drift in the time series, an average intensity projection can be created in ImageJ. Open the TIFF image stack in ImageJ and select the “Image → Stacks → Z project...” command. Select the “Average Intensity” option and click OK. In the newly generated time average image, CESA particles in the plasma membrane should appear as linear tracks. Background Golgi signal will appear as wide diffuse tracks and larger bright spots (Figure 3A).
- Occasionally, a horizontal drift may occur due to movement of the seedling or movement of the confocal stage. This can be detected by applying Z project on the movie, as the tracks will smear (Figure 3B demonstrates a Z stack of such event). We prefer not to analyze this type of movies, although in mild cases, the StackReg ImageJ plugin can be used.
- Observe the seedling under a confocal microscope featuring a spinning disk head mounted on a motorized inverted microscope with a 100x/1.4 numerical aperture oil immersion objective.
- Image processing
- Open the time-lapse series TIFF stack in ImageJ.
- As the images of the time-lapse series tend to be noisy and can vary in brightness depending on drift in the focal plane during acquisition, two filters can be applied in order to improve downstream analyses. The results of these operations are shown in Figure 4.
- First, select “Process → Enhance Contrast…” and set to “0.4%” saturated pixels. This saturation works well for our imaging. Select the option for “Process all X slices” and leave the remaining options unselected (Figure 4B).
- Noise can be greatly reduced by applying a walking average filter. Select “Plugins → Walking Average.” Enter “4” as the number of frames to average. Note that the resulting stack will contain 3 fewer frames than the initial stack (Figure 4C).
- Save the resulting stack and analyze particle velocity either by Kymograph in ImageJ or particle analysis in Imaris.
- Open the time-lapse series TIFF stack in ImageJ.
- Kymograph analysis in ImageJ
- Starting with the file generated by step D of “Image Processing,” create an average intensity projection as mentioned before.
- Use the straight line selection tool to trace a line that follows a CSC track in the time average image (Figure 5A).
- Switch back to the original time-lapse stack and select “Edit → Selection → Restore Selection.” The line drawn in step E2 should now be transferred to the time-lapse stack. (Figure 5B).
- Select “Plugins → MultipleKymograph.” Set the line width to “3”. This setting was shown to give the best result.
- A kymograph is generated depicting displacement along the defined path in the X dimension and time in the Y dimension (Figure 5C). Particles moving at a constant velocity thus appear as diagonal lines and their slope can be used to calculate the velocity of each particle in the kymograph.
- To easily determine the slope of a line, select “Analyze → Set Measurements…” and make sure that “Bounding Rectangle” is selected.
- On the kymograph, use the straight line tool to trace one of the diagonal lines corresponding to an individual CESA particle (Figure 5C).
- Press “M” to measure this line and the Results window will appear. Width and Height of the bounding box are the number of pixels corresponding to the displacement and time, respectively, that the particle has traveled.
- Calculating the velocity of each particle is straightforward, as each pixel in the Y dimension corresponds to the number of seconds between exposures in the time lapse, and each pixel in the X dimension has the same dimensions as the X and Y axes in the original image (This will vary depending on microscope and objective). For the path outlined in Figure 5C, where a frame was taken every 5 sec and each pixel in the original image corresponds to 135 nm:
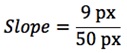
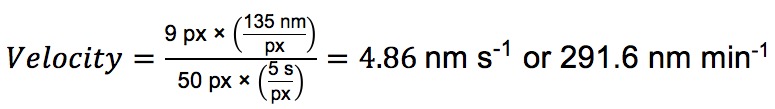
- Starting with the file generated by step D of “Image Processing,” create an average intensity projection as mentioned before.
- Imaris analysis of CSC velocity
- Imaris has a range of powerful tools for analyzing particle movement. A simple procedure for extracting plasma membrane CSC velocities while ignoring Golgi and other trafficking compartments is outlined here. These instructions are for Imaris 7.2 and may differ with later versions of the software.
- Ensure that the stack is saved as an 8 bit TIFF image after initial processing in ImageJ.
- Open the TIFF stack in Imaris “File → Open…”.
- Select “Image Processing → Swap Time and Z”.
- Select “Edit → Image Properties.” Set the voxel dimensions according to your microscope and objective. For example, with the microscope’s 100x objective used in this work, one pixel is 135 nm, so voxel size is 135 for X, 135 for Y and Z can be left as 1. Change the units to “nm.”
- In the same window, under the Time Point section, click “All Equidistant…” and enter the time interval used during acquisition. If the instructions at step C4 were followed, this is set to 5 sec. Click “OK.”
- Select “Surpass → Spots.” In the Spots tab, make sure that “Track Spots (over time)” is selected. In this example, the whole image will be processed, but a region of interest can be analyzed by selecting “Segment only a Region of Interest” and further deselecting “Process entire Image Finally”.
- Click the blue next arrow.
- Enter an estimated diameter for the spot detection. For this example, in our lab, 250 nm works well. You can measure several particles in Slice view to estimate particle size. Select “Background Subtraction.” Press the next button.
- Spots will be detected and will be marked with small squares as shown in Figure 6A. Quality thresholds can be adjusted at this point, but it is not critical to make sure that every spot is tracking a CESA particle. False positive spots can be filtered out by additional criteria later. Generally, adjust the threshold so that many spots are detected within cells, but few spots are detected in background regions. Click the next button.
- Click the next button, as there are no changes to be made on the Edit Spots screen.
- Select “Connected Components” and click next. This may take a couple of minutes.
- On the Classify Tracks screen, apply a filter of Track Duration above 60 sec. This will eliminate most of the fast moving trafficking compartments that are not associated with the plasma membrane. The remaining tracks are highlighted by color coded lines (Figure 6B).
- Click the green “finish” button.
- Click on the statistics tab, and then on the Detailed tab. Click the icon with multiple disks to export all statistics as a Microsoft Excel document.
- To calculate the velocity of the tracks, we take the Track Displacement Length and divide it by the Track Duration. The track speed that is calculated by Imaris is less accurate since at each time point the center of each particle is not determined reliably. Typically, 1,000 or more spots can be analyzed in a single movie.
- Imaris has a range of powerful tools for analyzing particle movement. A simple procedure for extracting plasma membrane CSC velocities while ignoring Golgi and other trafficking compartments is outlined here. These instructions are for Imaris 7.2 and may differ with later versions of the software.
Representative data

Figure 1. Sample preparation. A. Vacuum grease is applied at four corners in the slide. B. Arabidopsis seedling is transferred to the slide. C. Arabidopsis seedling is mounted with a cover glass. 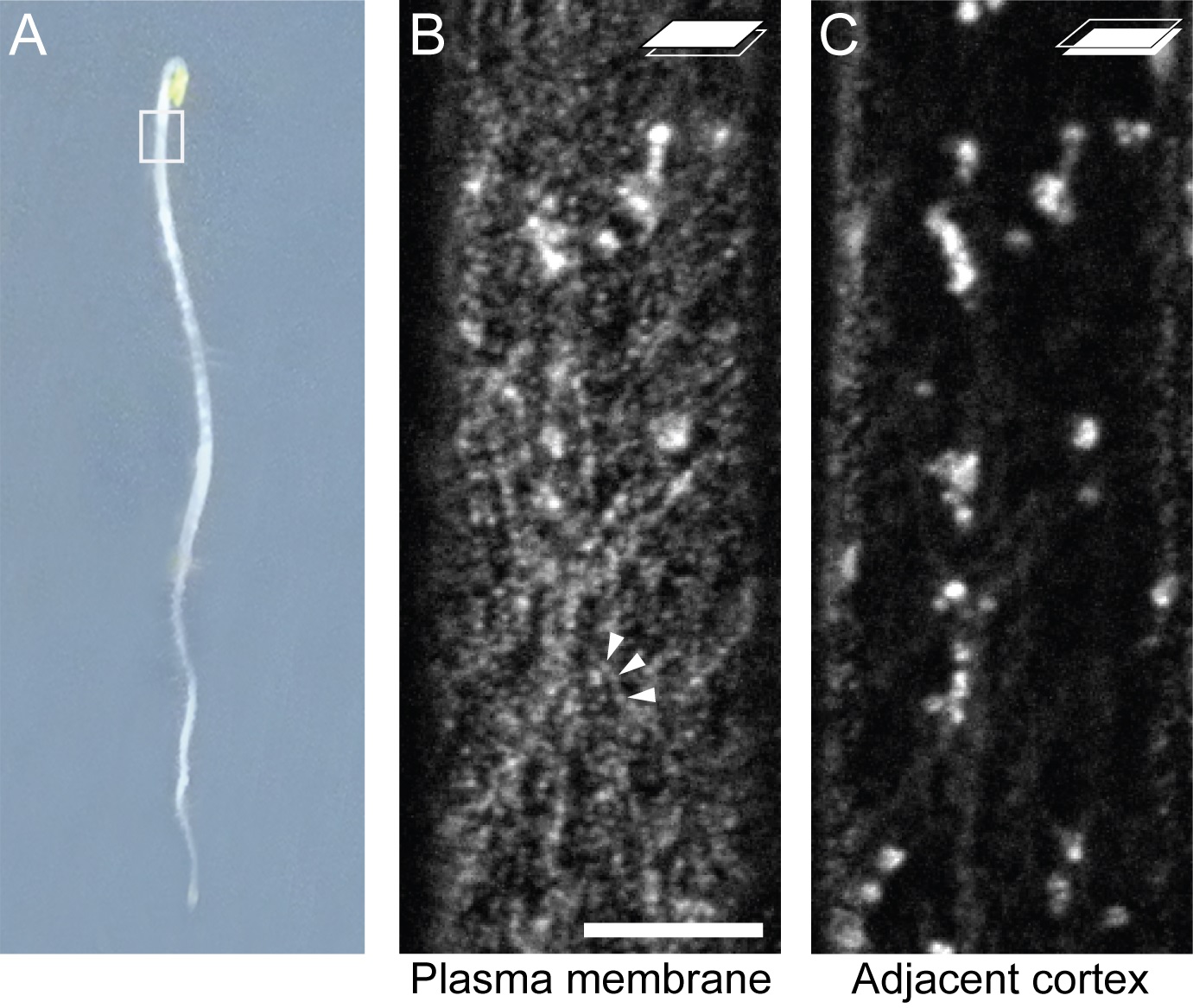
Figure 2. GFP:CESA3 localization in etiolated hypocotyl cells. Imaging was performed on epidermal cells in the upper hypocotyl from etiolated seedlings (A). Adjacent focal planes of the same cell showing GFP:CESA3 particles at the plasma membrane indicated by arrowheads (B), and Golgi and cytoplasmic compartment at lower cell cortex (C). Scale bar: 10 μm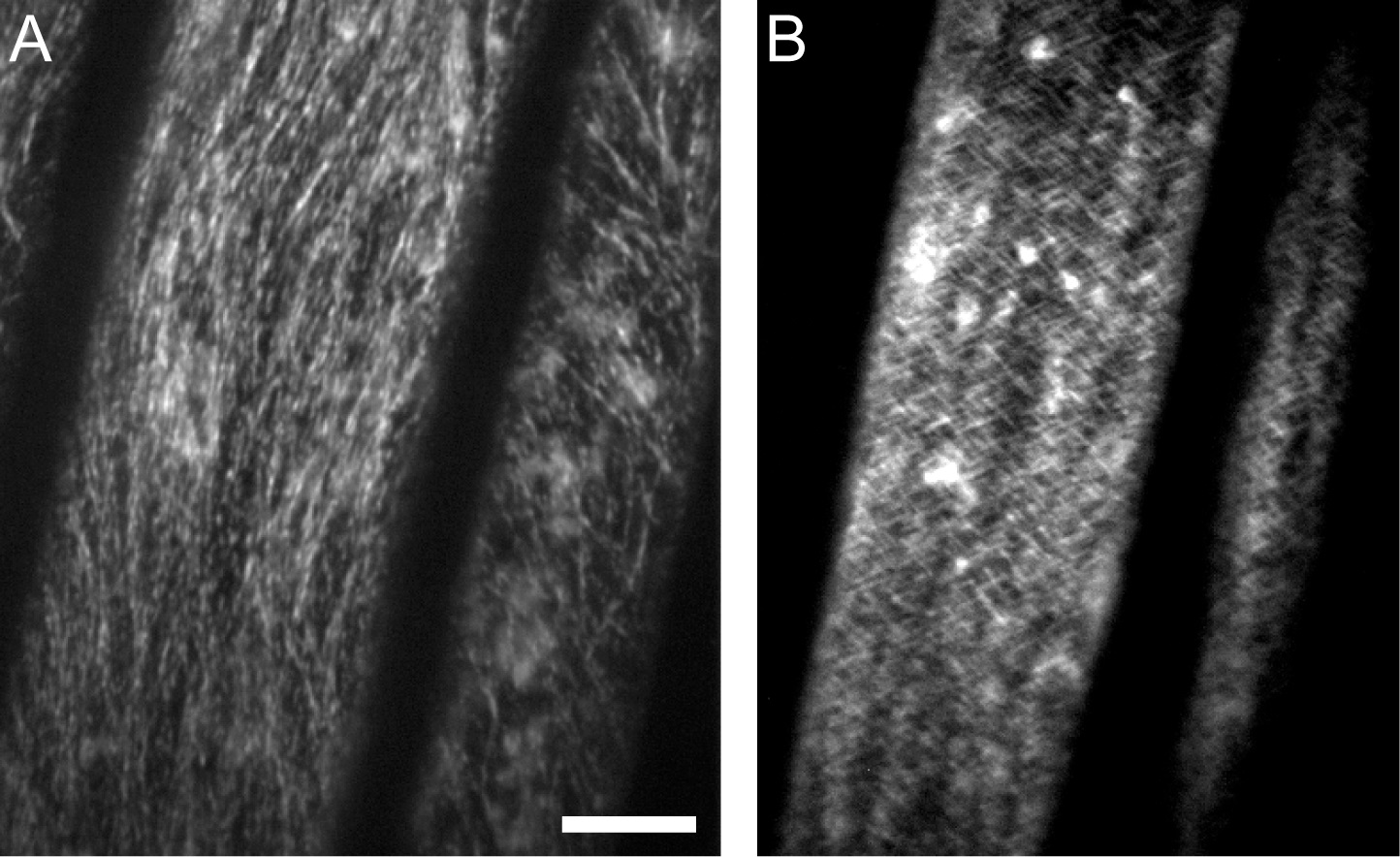
Figure 3. GFP-CESA3 time average images. A. An average intensity projection generated by ImageJ where CESA particles appear as linear tracks. B. An average intensity projection showing that the tracks smear when a horizontal stage drift takes place. Scale bar: 10 μm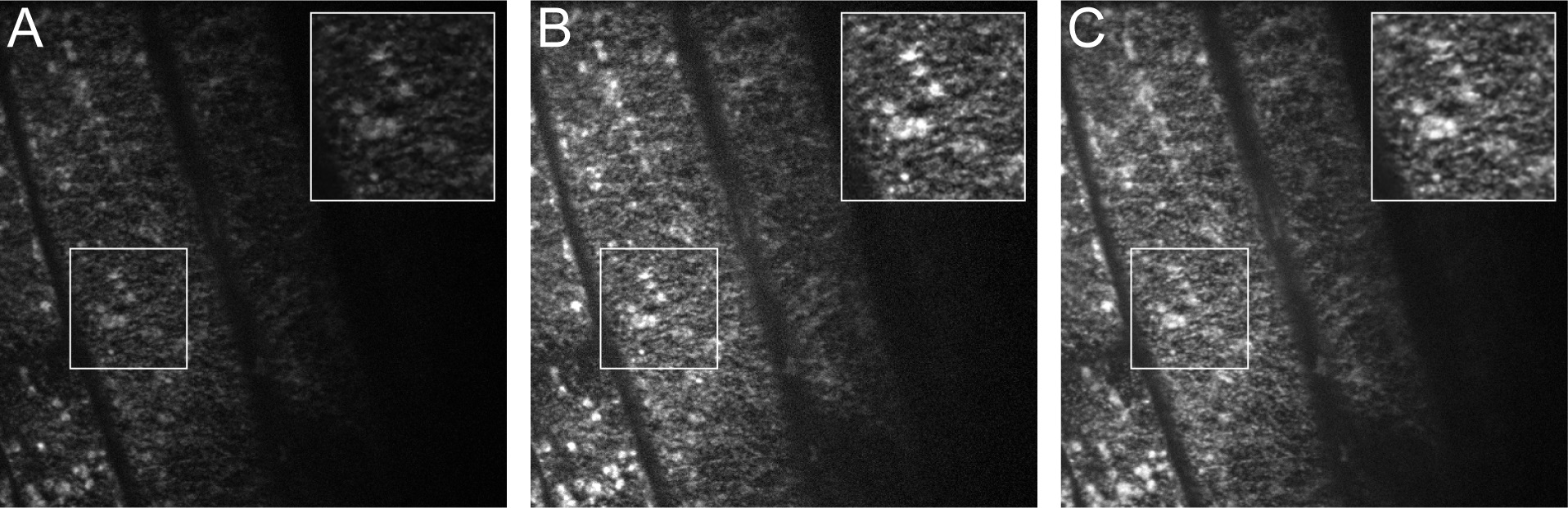
Figure 4. Image Processing in ImageJ. A. One frame from the original unprocessed stack. B. The same frame after “Enhance Contrast.” C. The same frame after “Enhance Contrast” and “Walking Average”. Insets represent an enlargement of the boxed region from the image.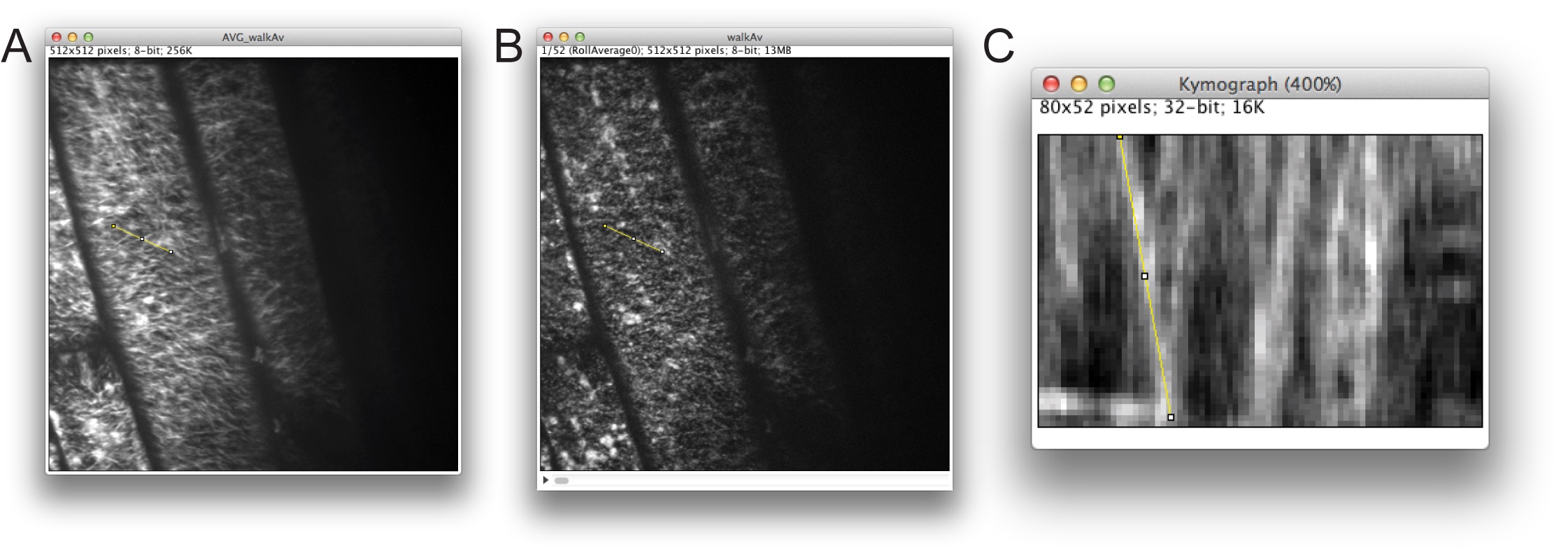
Figure 5. Defining a track for generation of a kymograph in ImageJ. A. Time average image used for selecting a linear track to analyze. B. The same selection transferred to the time-lapse stack. C. Kymograph analysis.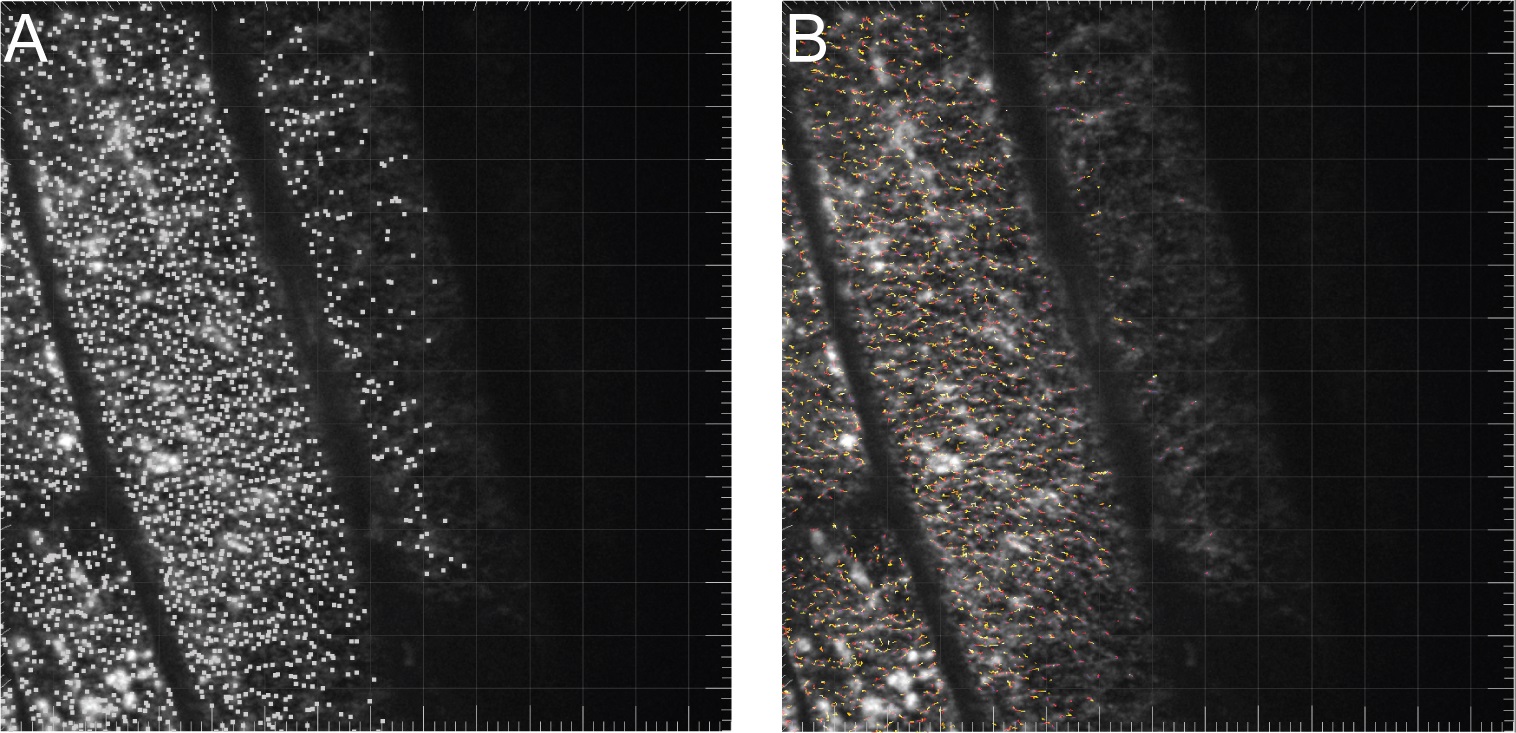
Figure 6. Analysis of particle movement using Imaris. A. Spots are detected and marked with small squares. B. Tracks after filtering based on duration of greater than 60 sec.
Notes
- Velocity of the CESA particles is variable depending on temperature and other factors; therefore comparative assays should be performed on the same day and under the same imaging conditions.
- No changes in complex motility were observed when seedlings were grown with or without sucrose in the plates.
- The N-terminal fusion of the fluorescent protein was required for all the constructs to be functional.
- All of these fluorescent protein fusions can be expressed either wild type or mutant backgrounds. It has been observed that GFP:CESA3 constructs are not able to rescue null alleles of CESA3, although hypomorphic alleles such as cesa3je5 are complemented (Desprez et al., 2007).
- Fluorescent marker YFP can also be excited at 514 nm if the laser is available.
Recipes
- 0.5x Murashige and Skoog (MS) media (1 L)
Add 2.2 g MS salts
Add 0.5 g MES
Add 10 g sucrose
Adjust to pH of 5.7 with 1 M KOH
Add dH2O to 1 L
Add 8 g agar for a solid media
Autoclave at 121 °C for 30 min
Acknowledgments
This protocol was adapted from the previously published studies, Paredez et al. (2006). We gratefully thank Prof. Chris Somerville, Dr. Heidi Szemenyei and Dr. Charles T. Anderson for critical reading of the protocol. This work was carried out in the laboratory of Chris Somerville at UC Berkeley. TV was supported by an EMBO long-term fellowship (ALTF 711-2012) and by postdoctoral funding from the Philomathia Foundation. TY was also supported by a fellowship from the Philomathia Foundation. NS was the recipient of Postdoctoral Award No. FI-434-2010 from the Binational Agricultural Research and Development Fund.
References
- Brabham, C. and Debolt, S. (2012). Chemical genetics to examine cellulose biosynthesis. Front Plant Sci 3: 309.
- Desprez, T., Juraniec, M., Crowell, E. F., Jouy, H., Pochylova, Z., Parcy, F., Hofte, H., Gonneau, M. and Vernhettes, S. (2007). Organization of cellulose synthase complexes involved in primary cell wall synthesis in Arabidopsis thaliana. Proc Natl Acad Sci U S A 104(39): 15572-15577.
- Paredez, A. R., Somerville, C. R. and Ehrhardt, D. W. (2006). Visualization of cellulose synthase demonstrates functional association with microtubules. Science 312(5779): 1491-1495.
- Sanchez-Rodriguez, C., Bauer, S., Hematy, K., Saxe, F., Ibanez, A. B., Vodermaier, V., Konlechner, C., Sampathkumar, A., Ruggeberg, M., Aichinger, E., Neumetzler, L., Burgert, I., Somerville, C., Hauser, M. T. and Persson, S. (2012). Chitinase-like1/pom-pom1 and its homolog CTL2 are glucan-interacting proteins important for cellulose biosynthesis in Arabidopsis. Plant Cell 24(2): 589-607.
- Sorek, N., Sorek, H., Kijac, A., Szemenyei, H. J., Bauer, S., Hematy, K., Wemmer, D. E. and Somerville, C. R. (2014). The Arabidopsis COBRA protein facilitates cellulose crystallization at the plasma membrane. J Biol Chem 289(50): 34911-34920.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Vellosillo, T., Yeats, T. and Sorek, N. (2015). Analysis of in vivo Cellulose Biosynthesis in Arabidopsis Cells by Spinning Disk Confocal Microscopy. Bio-protocol 5(19): e1617. DOI: 10.21769/BioProtoc.1617.
Category
Plant Science > Plant biochemistry > Carbohydrate
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.