- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for the Generation of a Transcription Factor Open Reading Frame Collection (TFome)
(*contributed equally to this work) Published: Vol 5, Iss 15, Aug 5, 2015 DOI: 10.21769/BioProtoc.1547 Views: 16280
Reviewed by: Arsalan DaudiVinay PanwarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The construction of a physical collection of open reading frames (ORFeomes) for genes of any model organism is a useful tool for the exploration of gene function, gene regulation, and protein-protein interaction. Here we describe in detail a protocol that has been used to develop the first collection of transcription factor (TF) and co-regulator (CR) open reading frames (TFome) in maize (Burdo et al., 2014). This TFome is being used to establish the architecture of gene regulatory networks (GRNs) responsible for the control of transcription of all genes in an organism. The protocol outlined here describes how to proceed when only an incomplete genome with partial annotation is available. TFome clones are made in a recombination-ready vector of the Gateway? system, allowing for the facile transfer of the ORFs to other Gateway?-compatible vectors, such as those suitable for expression in other host species. Although this protocol was developed for the maize TFome it can readily be applied to the generation of complete ORFeome collections in other eukaryotic species.
[Protocol overview] An important aspect of successful TFome generation is the initial effort spent to establish a reliable set of gene models so that they can be subsequently amplified or synthesized. An actual TFome construction protocol for a particular species will depend on available resources such as a full-length cDNA (flcDNA) collection and a reliable reference genome (Figure 1).
In the case of maize, a flcDNA collection and a draft genome was available, but the former provided only 30% of the needed clones, and the latter contained gaps and some erroneous gene models. In order to develop a near-complete set of target gene models for maize TFs, a bioinformatics pipeline was developed as described by Yilmaz et al. (2009). In brief, a two-pronged search process was developed. The first involved making a collection of protein sequences of TFs in other species and available from databases such as PlantTFDB, PlnTFDB and DBDTF. These sequences were then used to search gene models from the draft maize genome using BLASTP. The second process involved developing a collection of domains that define TF families and that are mostly annotated in the PFAM database (Finn et al., 2014). These domains were then used to search the draft maize genome using BLASTX. The number of TF families that exist and their naming is subject to change as new members are discovered and studied. Table 1 provides a list of known TF families with alternative names along with the respective PFAM domains whose presence or absence defines each TF family. HMM models for each domain can be obtained from the PFAM database (pfam.xfam.org). Following the BLAST search, redundant models are eliminated and then based on the TF motifs present in each gene model, gene models are assigned to a TF or Co-Regulator (CR) family according to the criteria specified in Table 1. Lastly, it is recommended to set up a database to store information on each TF family. The GRASSIUS (www.grassius.org) website was established to access the stored information on TF gene models for maize, sorghum, rice, Brachypodium, sugarcane and other grasses (Burdo et al., 2014). In the following section, an assumption is made that at least a draft genome or draft transcriptome is available and that a set of gene models is available that have been determined ab initio or with additional manual annotation. Familiarity with the use of PERL scripts is advantageous for the gene model assembly phase.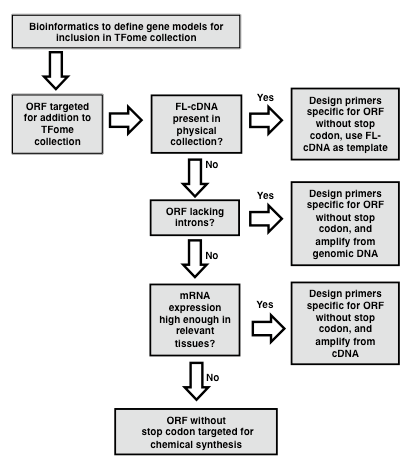
Figure 1. Flowchart for the generation of a TFome project. Flowchart outlining the general strategy for template identification, PCR amplification and cloning of transcription factor (TF) full length (FL) open reading frames (ORFs). (modified from Burdo et al., 2014)
Materials and Reagents
The main starting materials for embarking upon a large TFome project are the assembly of gene models and a collection of plasmid templates from existing cDNA collections. In this section, processes to develop these are outlined.
- Assembly of gene models and TF domain databases
- Assemble a collection of gene models from the available target genome. For plant genomes, the Phytozome database (phytozome.jgi.doe.gov) and EnsemblPlants (http://plants.ensembl.org/index.html) permit the downloading of all predicted protein models for a species as a single multi-sequence FASTA formatted file. For example, at the Phytozome website the BioMart tool permits one to select the genome dataset for a particular plant species. Once the genome is selected then attributes of the sequences to be downloaded can be specified such as peptide sequences or coding sequences only. By default all gene models are selected and then these can be downloaded as a single FASTA file that is the input file for subsequent steps below.
- Assuming the draft quality of the transcript annotation it would be desirable to eliminate redundant gene models in a multiseq FASTA file. The GRASSIUS website provides the custom perl script “IdentifySeqRedundancy.pl” (http://grassius.org/tools.html) for this purpose. This script also requires the perl module “Digest::MD5” which is available from the Comprehensive Perl Archive Network (CPAN) website (http://search.cpan.org/dist/Digest-MD5/MD5.pm). One can also eliminate redundant models within the species using BLAST searches. Proteins were arbitrarily considered duplicated if they are found in the same species, with a query coverage ≥ 90%, or have a query identity ≥ 90% and the query alignment starts less than 9 residues from the start codon. Alternatively, more complex criteria such as those of Gu et al., 2002, may be employed to identify duplicate proteins in a genome. If these conditions are satisfied, the longest protein is kept and the eliminated proteins were classified as identical or splicing variants. If there is access to RNA-Seq datasets they may be used to corroborate target TF gene models, but such an approach is not outlined here. Targeting the longest splice variant which is supported by EST or RNA-Seq data provides the maximum protein interaction space for identifying the protein-DNA and protein-protein interactions that gene regulatory networks are comprised of.
- Scanning the non-redundant multifasta file against a collection of protein domains as hidden Markov models (HMMs) such as provided by PFAM or Interpro provides a protein domain annotation of the putative proteome. For this particular step the software HMMER is required. In brief HMMER is a set of tools implementing the profile hidden Markov models to find similarity across protein sequences and is necessary to search the PFAM HMM models in a group of protein sequences. It is possible to call HMMER from a variety of scripting languages such as perl or python, using pre-build HMMER wrappers. PFAM provides one of those scripts written in perl (pfamscan.pl) with a set of PFAM pre-established parameters. Source code may be downloaded from the following website ftp://ftp.sanger.ac.uk/pub/databases/Pfam/Tools/PfamScan.tar.gz. Table 1 is a compilation of protein domains that have been used to define TF and CR families in plants (Buitrago-Florez et al., 2014; Burdo et al., 2014; Perez-Rodriguez et al., 2010; Yilmaz et al., 2009). This table describes 62 TF and 26 CR families that were used in defining gene models for inclusion in the maize TFome. Twelve of these families do not have ascribed PFAM domains but can be defined using “in house” HMMs as previously described (Buitrago-Florez et al., 2014; Perez-Rodriguez et al., 2010). The HMMs for the CCAAT-Hap2, 3, and 5 TF families can be obtained from the AGRIS database (http://arabidopsis.med.ohio-state.edu) (Yilmaz et al., 2011).
- Once the primary list of gene models are annotated with protein domains, they are assigned to TF or CR families using the criteria outlined in Table 1. The criteria include identifying the presence of one or more motifs and the absence of other (forbidden) motifs (Buitrago-Florez et al., 2014; Perez-Rodriguez et al., 2010; Yilmaz et al., 2009). The annotation involves a custom perl script “get_InterProScanDomains.pl” to sort proteins into TF families based on the interproscan output and is available at the GRASSIUS website (http://grassius.org/tools.html). The script keeps only significant hits with e-value ≤ 0.001 and verifies that the rules as defined in Table 1 are fulfilled. When the rules are only partially fulfilled then the protein is assigned into “Orphan” TFs. This can be the largest class of TFs for a species (~11.7% of the maize TFome), until Orphan members are assigned to families.
- Searches can be conducted with local computation resources or when not available using the iPlant Collaborative Discovery Environment public resource (http://www.iplantcollaborative.org).
- Assemble a collection of gene models from the available target genome. For plant genomes, the Phytozome database (phytozome.jgi.doe.gov) and EnsemblPlants (http://plants.ensembl.org/index.html) permit the downloading of all predicted protein models for a species as a single multi-sequence FASTA formatted file. For example, at the Phytozome website the BioMart tool permits one to select the genome dataset for a particular plant species. Once the genome is selected then attributes of the sequences to be downloaded can be specified such as peptide sequences or coding sequences only. By default all gene models are selected and then these can be downloaded as a single FASTA file that is the input file for subsequent steps below.
- Screening of EST or flcDNA collections for plasmid templates
- Identify which cDNA resources are available for the target genome (see Note 1). Most genome projects for model species include generating an EST library, although many libraries are not publically available. For the maize TFome, extensive use of the maize flcDNA collection (http://www.maizecdna.org) was made (Soderlund et al., 2009). Individual cDNA clones for this library and many other plant species are available through the Arizona Genomics Institute (http://www.genome.arizona.edu).
- Using the coding sequence of a gene model as a query sequence, ESTs or flcDNAs were then identified using BLASTP or BLASTX, for which the amino terminus, including the start codon, was present and the sequence alignment was >99%. Alignments that are not 100% identical may occur because EST sequences are not high fidelity due to sequencing errors particularly at the 3’ end of sequences (Soderlund et al., 2009). Alternatively spliced isoforms not represented by a transcript model may also be targeted, as long as they maintain the reading frame of the original transcript.
- Once a likely flcDNA template is located, then the plasmid is acquired, isolated and sequenced from each end to confirm that: a) it is the correct template, and b) it is full length. Some cDNA libraries utilize enzymes during their construction that cut within the coding sequence leading to partial clones, which would be unsuitable for amplification of the entire coding sequence.
- Once a suitable target plasmid template is identified for a target gene, then a sample is diluted to 1 ng/µl and 1 ng is sufficient as template in a PCR reaction.
- Identify which cDNA resources are available for the target genome (see Note 1). Most genome projects for model species include generating an EST library, although many libraries are not publically available. For the maize TFome, extensive use of the maize flcDNA collection (http://www.maizecdna.org) was made (Soderlund et al., 2009). Individual cDNA clones for this library and many other plant species are available through the Arizona Genomics Institute (http://www.genome.arizona.edu).
- Other reagents
- One Shot® TOP10 chemically competent cells (Life Technologies, catalog number: C404003 )
- PureLink® Plant RNA Reagent (Life Technologies, catalog number: 2322-012 )
- Turbo DNA-free™ kit (Life Technologies, Ambion®, catalog number: AM1907 )
- Thermo Scientific Maxima H minus 1st strand synthesis kit (Thermo Fisher Scientific, catalog number: K1652 )
- Ribolock® RNase inhibitor (Thermo Fisher Scientific, catalog number: EO0382 )
- EmeraldAmp® MAX PCR Master Mix (Takara Bio Company, catalog number: RR320A )
- Phusion® high fidelity polymerase (New England Biolabs, catalog number: M0530S )
- Gateway® pENTR™/D-TOPO® or pENTR™/SD/D-TOPO® vectors (Life Technologies, catalog numbers: K243520 and K242020 respectively)
- Genscript® Taq Polymerase (GenScript USA Inc., catalog number: E000071000 )
- EmeraldAmp® MAX PCR Master Mix (Takara Bio Company, catalog number: RR320A)
- 1 kb ladder molecular weight size standards (Thermo Fisher Scientific, catalog number: SM0311 )
- RNA extraction buffer I (see Recipes)
- RNA extraction buffer II stock solutions (see Recipes)
- RNA extraction buffer II (working solution) (see Recipes)
- Carlson lysis buffer (see Recipes)
- Freezing medium (see Recipes)
- One Shot® TOP10 chemically competent cells (Life Technologies, catalog number: C404003 )
Equipment
Note: Most equipment listed here is standard molecular biology instrumentation present in most laboratories. A large TFome project would also benefit from the use of multichannel pippetters and 96 well plate formatted experimentation.
- Microcentrifuge
- High speed centrifuge
- Gradient thermocycler
- Gel electrophoresis equipment
- Water baths
- -80 °C freezer for the storage of stocks
- Aerosol pipette tips recommended for all DNA manipulation and cloning experiments
- Wizard® SV 96 Plasmid DNA Purification System (Promega Corporation, catalog number: A2255 )
- Wizard® Plus SV Minipreps DNA Purification System (Promega Corporation, catalog number: A1330 )
- Wizard® SV Gel and PCR Clean-Up System (Promega Corporation, catalog number: A9281 )
- Gene synthesis (Life Technologies)
- 2 ml 96 well culture dish (Thermo Fisher Scientific, catalog number: 12566121 )
- Breathable plate seal (Thermo Fisher Scientific, catalog number: AB0718 )
- 0.5 ml plates (USA Scientific, catalog number: 18965000 )
- 7 mm sized silicone seal that can be re-autoclaved (Thermo Fisher Scientific, catalog number: 0339649 )
- Matrix sample storage system (matrixtechcorp.com, Thermo Fisher Scientific, catalog numbers: 4111MAT ) (Capit-All® Capper/Decapper), 3740 (Barcoded screw cap tubes), and 4477 (Screw cap tray)
- (Optional) Vac-Man® 96 Vacuum Manifold (Promega Corporation, catalog number: A2291 ) for 96 well plasmid preparation
- (Optional) Vacuum pump capable of generating 38-51 cm of Hg or equivalent
Software
- OligoAnalyzer 3.1 software (www.idtdna.com)
Procedure
- Preparation of nucleic acid templates for PCR
Here, three main sources of template for the amplification of TF coding sequences were used: 1) full length cDNA clones from representative cDNA libraries, 2) cDNA generated from RNA purified from a variety of host species tissues, and 3) genomic DNA from the host species which is suitable for the amplification of coding sequences from single exon genes. Methods for the preparation of nucleic acid templates are provided in this section.- Plasmid templates for TF ORF amplification.
Any traditional plasmid preparation will provide plasmid of sufficient quantity and purity to act as template for PCR amplification. For the maize TFome project, hundreds of plasmids were required and a 96-well format was desirable. The high throughput system that proved satisfactory was the Wizard® SV 96 Plasmid DNA Purification System. This system requires a vacuum pump capable of generating 38-51 cm of Hg or equivalent and the Vac-Man® 96 Vacuum Manifold. It is noted however that the final concentration of plasmid DNA from this method was usually less than the minimum required for automated Sanger dideoxy DNA sequencing which is approximately 50 ng/μl. If the plasmid template needs to be verified by DNA sequencing prior to amplification then a higher yielding protocol will be required (e. g. Wizard® Plus SV Minipreps DNA Purification System). - cDNA template for TF ORF amplification by Reverse Transcription-PCR (RT-PCR).
Most TF genes are restricted in their temporal and tissue specific gene expression pattern. When considering the use of RT-PCR for the generation of amplicons it is helpful to have some evidence for the tissue in which a particular TF is most highly expressed to maximize success in amplification. For maize and a few other plant species, the qTeller website (www.qteller.com) provides a searchable resource that displays the relative abundance of transcripts of genes from RNA-Seq data. For maize genes that were successfully amplified, it was found that they were always amplified from one of the top three ranked tissues where their expression was deemed highest using the qTeller resource. The Bio-Analytic Resource for Plant Biology (BAR) (www.bar.utoronto.ca) and The Plant Expression (PLEXDB) (http://www.plexdb.org) databases provide similar resources for a broader selection of plant species.
For the collection of RNA samples, a large plant population (about 100 maize plants were employed) should be planted since multiple tissues will be required and the collection of certain tissues (e.g. developing ears) will entail harvesting the entire plant. For some genes, biotic and abiotic stress, or circadian rhythm, may cause transcript levels to increase significantly. In the case of the maize TFome, it was found that more than half of the clones derived by RT-PCR could be amplified from young seedling shoots and roots (Burdo et al., 2014).
RNA isolation from plant tissues
Total RNA was isolated from plant tissues by one of two methods. For non-starchy tissues PureLink® Plant RNA Reagent was employed according to the manufacturer's recommendations. For the isolation of RNA from starchy tissues, such as developing and mature seeds, the method described by Li and colleagues (Li and Trick, 2005) was utilized with slight modifications. For convenience, the protocol and solution recipes are detailed below. Typically, total RNA was isolated from 1 gram of tissue.- Grind 1 gram of tissue in mortar and pestle with liquid nitrogen. For successful implementation, it is essential that tissues are immediately frozen in liquid nitrogen at harvest time and stored at -80 °C before grinding. The tissue should not be permitted to thaw at any time prior to the addition of extraction buffer.
- Transfer powder into RNase free centrifuge tube with 4 ml of extraction buffer I and immediately mix vigorously.
- Add 2.5 ml of phenol:chloroform mixture (1:1, pH 4.7) and mix well by inversion.
- Centrifuge at 13,000 x g for 15 in at 4 °C.
- Transfer upper aqueous phase (2.5 ml) to a new RNase free centrifuge tube containing 2.5 ml extraction buffer II. Samples are mixed by gentle inversion and incubated at room temperature for 10 min.
- Add 2 ml of chloroform-isoamyl alcohol (24:1), mix well and centrifuge the samples at 13,000 x g for 15 min at 4 °C.
- Recover supernatants (4.5 ml), and add 3 ml of isopropanol and 2.5 ml of 1.2 M NaCl. Samples are mixed by gentle inversion, incubated on ice for 15 min, and centrifuged at 13,000 x g for 15 min at 4 °C.
- Discard the supernatants and wash the pellets carefully with 400 µl of 70% ethanol.
- Dry the washed pellets at room temperature and resuspend in RNase free water (about 500 µl) prior to storing at -70 °C.
Typically yields of total RNA were 2.2 ± 0.9 µg/µl with A260/A280 and A260/A230 ratios of 1.93 ± 0.06 and 2.32 ± 0.32 respectively (n = 30). Total RNA samples with ratios <1.7 and 2.0 were deemed unsuitable. RNA integrity must be assessed by separation on a denaturing gel. The appearance of acceptable quality total RNA is shown in Figure 2A. For most tissues, the large and small ribosomal bands should be sharp with the upper band being twice the intensity of the lower band. A faint smear extending above and below the ribosomal bands is indicative of mRNA. For germinating seeds, the ribosomal bands will have diminished intensity but the smear should still be visible. Some large molecular weight material is indicative of contaminating DNA, which is removed by DNase treatment (see below).
Isolation of genomic DNA
For TF genes whose coding sequence is contained within one exon (about 7% of TF genes in maize), then total genomic DNA is a suitable template for amplicon generation. The source material should match the reference genome that is being employed in the project. This will allow the user to discern if single nucleotide polymorphisms (SNPs) in the amplicons are due to errors during amplification and not due to naturally occurring SNPs in the germplasm. There are many suitable genomic DNA isolation protocols available and the one used in this project is summarized below.
Plant DNA isolation protocol (modified from Carlson et al., 1991).- Grind 1 g of leaf material in liquid nitrogen with a cold mortar and pestle.
- Transfer ground material into a sterile Oakridge tube containing 10 ml of Carlson Buffer preheated to 70 °C (by standing in water bath).
- Incubate for 20 min at 70 °C by standing in water bath - invert every few minutes to mix contents (use a shaking water bath if available).
- Cool samples to room temperature and add 5 ml of chloroform/isoamyl alcohol (24:1) and vortex well for 20 sec. Then centrifuge for 10 min at 4 °C at 5,000 x g.
- Transfer upper aqueous phase into a fresh (sterile) Oakridge tube. Add 50 µl of 10 mg/ml RNase A (preboiled and frozen at -20 °C) - incubate at 37 °C for 20 min to degrade RNA.
- Add an equal volume of isopropanol (2-propanol), vortex and incubate at room temperature for 20 min to allow the precipitation of DNA. Centrifuge for 20 min at 4 °C at 5,000 x g.
- Resuspend the pellet in 600 µl of TE (1 mM Tris, 0.1 mM EDTA, pH 8.0) and transfer into an Eppendorf tube.
- Add 5 µl of 10 mg/ml RNase A and incubate for 30 min at 37 °C.
- Extract the DNA with an equal volume of phenol/chloroform/isoamylalcohol (25:24:1) and centrifuge at 14,000 x g for 5 min at 4 °C.
- Transfer the top aqueous phase to a new Eppendorf tube. Extract with an equal volume (now about 0.5 ml) of chloroform/isoamylaclcohol (24:1) and centrifuge at 14,000 x g for 5 min in a benchtop microcentrifuge (4 °C).
- Precipitate DNA by adding 1/10 volume of 3 M NaOAc or 5 M NH4OAc and 2 volumes of 100% ethanol. Mix by gentle inversion and centrifuge at 14,000 x g for 5 min at 4 °C.
- Wash the DNA pellet with 70% ethanol once or twice.
- Remove the wash supernatant and let the pellet air dry for about 10 min but it is best not to let the DNA become completely dry. Resuspend the DNA in about 200 µl of TE buffer (1 mM Tris, 0.1 mM EDTA, pH 8.0) and determine the concentration by UV spectroscopy.
DNA isolated by this method should have an A260/A280 ratio > 1.7 and an A260/A230 ratio > 2.0. For a complex genome such as maize 100 ng should be used as template in a PCR reaction.
- Grind 1 gram of tissue in mortar and pestle with liquid nitrogen. For successful implementation, it is essential that tissues are immediately frozen in liquid nitrogen at harvest time and stored at -80 °C before grinding. The tissue should not be permitted to thaw at any time prior to the addition of extraction buffer.
- Plasmid templates for TF ORF amplification.
- Generation of complementary DNA (cDNA) from total RNA
- Prior to the generation of cDNA it is important to remove any contaminating genomic DNA from the total RNA preparations. In this project the Turbo DNA-free™ kit was employed according to the manufacturer's recommendations. Four units of TURBO™ DNase are used to remove any contaminating DNA from 20 µg of total RNA. Following treatment at 37 °C for 30 min, the DNase deactivation matrix is added at room temperature and after 5 min incubation, separated by centrifugation at 10,000 x g. The supernatant is removed and used in a reverse transcription reaction.
- It is recommended that a high fidelity reverse transcriptase is employed to reduce errors in the cDNA that will act as template for target gene amplification. We employed Thermo Scientific Maxima H minus 1st strand synthesis kit according to the manufacturer's recommendations except that 40 units of Ribolock® RNase inhibitor was added in each reverse transcription reaction. Using this protocol, 12 µl of DNase treated RNA (approximately 5 µg total RNA) is annealed to the provided oligo dT primer in a 20 µl reaction. Between 0.5 to 1 µl of this reaction is sufficient for a single PCR amplification of most TF genes. Before proceeding with PCR, quality of the cDNA was assessed by amplifying the transcript of a housekeeping gene. For this purpose, low fidelity DNA polymerase such as the EmeraldAmp® MAX PCR Master Mix is adequate. For maize, a 1,001 bp portion of the ZmGAPDH (GRMZM2G046804) transcript was amplified using the primers ZmGAPDH_F (5’-ATGCAGGCAAGATTAAGATCGGAATCAAC-3’) and ZmGAPDH_R (5’-CATGTGGCGGATCAGGTCGAC-3’). The absence of a larger 2,817 bp PCR product confirmed the removal of genomic DNA (Figure 2B).
- Prior to the generation of cDNA it is important to remove any contaminating genomic DNA from the total RNA preparations. In this project the Turbo DNA-free™ kit was employed according to the manufacturer's recommendations. Four units of TURBO™ DNase are used to remove any contaminating DNA from 20 µg of total RNA. Following treatment at 37 °C for 30 min, the DNase deactivation matrix is added at room temperature and after 5 min incubation, separated by centrifugation at 10,000 x g. The supernatant is removed and used in a reverse transcription reaction.
- Amplification of TF coding sequences
- Primers are synthesized to amplify the corresponding ORFs without the respective stop codons, and an additional 5'-CACC-3’ nucleotide tail added to the forward primer, for directional cloning into the pENTR™ vectors. For maize, 1,273 primer pairs were analyzed in regards to GC content and melting temperature (Tm). Tm values were estimated using the OligoAnalyzer 3.1 software (www.idtdna.com). The forward primers had an average length of 25 ± 4 bp including the 5’ CACC tail, which was similar to that of the reverse primers (23 ± 4 bp). However the GC content of the forward primers (62.8 ± 9.1) was about 12% higher than that of the reverse primer (50.2 ± 11.7). As a result the average Tm for the forward primer was about 3 °C higher (66 ± 5 °C) than that of the reverse primer (63 ± 5 °C). In addition the average GC content of the amplicons was 62.4 ± 8.8 % with a range from 78.9 to 38.2% (n = 1,411). Due to the relatively high GC content the buffer for GC rich templates was routinely employed for CDS amplification. When designing primers an effort should be made to have the difference in Tm (ΔTm) for primers in a pair to be as low as possible. In this project the average ΔTm was 3.2 ± 4.1 °C however amplicons were successfully obtained even when the ΔTm was as much as 19 °C.
- PCR products are next generated using a high-fidelity polymerase. For this project, the Phusion® high fidelity polymerase was chosen because it exhibits an error rate more than 50-fold lower than that of Taq DNA Polymerase. In addition, Phusion® polymerase has an enhanced processivity domain that requires an extension time of only about 15 sec per kb, and we found that an extension time of 1 min was sufficient for even the longest amplicons. The coding sequences of TFs in the maize genome were on average 1,024 ± 474 bp in length with a range from 90 to 3,705 bp. The recommended initial composition for PCR are listed in Table 2. It was determined that the use of GC buffer and 10% glycerol was the most successful initial condition (Table 2, setup A). If this initial condition was unsuccessful it is recommended to next adjust the annealing temperature (Note 2). It was found that the addition of DMSO or extra MgCl2 also improved the success rate for some templates (Table 2; Setups B and C). The cycling parameters for PCR are as follows: A single initial denaturation cycle of 98 °C for 1 min, 25- to 35 cycles of amplification of 98 °C denaturation for 20 sec, ~ 60 °C annealing for 40 sec, 72 °C extension for 15 sec per kb, 1 final extension step of 72 °C for 5 min. It is important that following completion of the final extension step that the PCR reactions be removed and processed or stored at -20 °C until processing (Note 3). The number of cycles depends on the template source. As few as 25 cycles may be employed to amplify from plasmid templates but usually 35 cycles are required when using a cDNA template. In general, a fewer number of cycles will reduce the chances of errors due to polymerase.
Table 2. Recommended PCR composition*Template amounts, Plasmid -1 ng, cDNA - 0.5 µl of RT reaction, genomic DNA-100 ngOptions Components (add in this order) A B C vol (µl) vol (µl) vol (µl) H2O 11 15.75 -15 10 - 10.75 GC Buffer (5x) contains 7.5 mM MgCl2 5 5 5 Forward Primer (20 µM stock) 1 1 1 Reverse Primer (20 µM stock) 1 1 1 dNTPs (25 mM stock) 1 1 1 MgCl2 (50 mM stock) 0 0 0.25 -1.0 Glycerol (50% stock) 5 0 5 DMSO (100% stock) 0 0.25 - 1.0 0 Template* (plasmid, cDNA, or DNA) 0.5 0.5 0.5 Phusion® Polymerase* (5 U/µl) 0.5 0.5 0.5 Total Volume 25 µl 25 µl 25 µl - Once the PCR is complete the products are separated by gel electrophoresis to identify products of the expected size. When plasmid templates are employed it is usual to observe a single band, however when genomic DNA or cDNA is employed or when longer fragments are expected it is more usual to observe multiple bands (Figure 2C). The presence of a band of the correct size is not a guarantee that the correct coding sequence has been amplified however especially for genes that belong to a TF family for which multiple paralogs exist.
- Primers are synthesized to amplify the corresponding ORFs without the respective stop codons, and an additional 5'-CACC-3’ nucleotide tail added to the forward primer, for directional cloning into the pENTR™ vectors. For maize, 1,273 primer pairs were analyzed in regards to GC content and melting temperature (Tm). Tm values were estimated using the OligoAnalyzer 3.1 software (www.idtdna.com). The forward primers had an average length of 25 ± 4 bp including the 5’ CACC tail, which was similar to that of the reverse primers (23 ± 4 bp). However the GC content of the forward primers (62.8 ± 9.1) was about 12% higher than that of the reverse primer (50.2 ± 11.7). As a result the average Tm for the forward primer was about 3 °C higher (66 ± 5 °C) than that of the reverse primer (63 ± 5 °C). In addition the average GC content of the amplicons was 62.4 ± 8.8 % with a range from 78.9 to 38.2% (n = 1,411). Due to the relatively high GC content the buffer for GC rich templates was routinely employed for CDS amplification. When designing primers an effort should be made to have the difference in Tm (ΔTm) for primers in a pair to be as low as possible. In this project the average ΔTm was 3.2 ± 4.1 °C however amplicons were successfully obtained even when the ΔTm was as much as 19 °C.
- Cloning and confirmation of PCR amplicons
- DNA bands of the correct size is identified by DNA gel electrophoresis are excised and gel purified. There are many suitable kits for purification for this step. For this project we used the Wizard® SV Gel and PCR Clean-Up System. Carefully quantify the purified product prior to ligation.
- Purified PCR products are then cloned into the Gateway® pENTR™/D-TOPO® or pENTR™/SD/D-TOPO® vectors. These vectors allow for the facile transfer of the clones into alternative expression vectors and greatly increase the utility of the TFome collection. Ligations were performed according to the manufacturers recommended protocol and used to transform One Shot® TOP 10 chemically competent cells. If desired half the reaction size can be performed using the recommended ratio of insert to vector.
- Kanamycin-resistant colonies were then screened for inserts of the correct size and orientation by colony PCR or by restriction digestion of candidate plasmids. We noted considerable variation in the cloning efficiency using the Gateway® entry cloning kits. This difficulty was overcome by using an entire kit at one time to maximize efficiency. If cloning efficiency is high then plasmid DNA may be isolated from one or two colonies and these used for confirmation. It was found however that due to variation in cloning efficiencies, that colony PCR is often a more efficient way of identifying inserts.
Colony PCR to identify positive clones
First a master mix is prepared of a general purpose DNA polymerase such as Genscript® Taq Polymerase, in the manufacturer provided buffer (50 mM KCl, 10 mM Tris HCl, pH 9.0 at 25 °C), 1.5 mM MgCl2, 1% Triton X-100). The EmeraldAmp® MAX PCR Master Mix was also found suitable for colony PCR). Between 5-10% DMSO may also be added. To this mixture, the appropriate forward and reverse primers are added to a final concentration of 0.4 µM. Then 25 µl aliquots are added to the well of individual PCR tubes or a 96 well PCR plate. A sterile toothpick or pipette tip is then used to isolate a single reference colony, that is first mixed into an individual PCR reaction tube, and then to plate a replica of the colony being screened. The cycling parameters for PCR are as follows: A single initial denaturation cycle of 95 °C for 5 min, 35 cycles of amplification of 95 °C denaturation for 1 min, ~ 60 °C annealing for 1 min, 72 °C extension for 1 min per kb. Following PCR the products are separated by gel electrophoresis and examined for a product of the expected size (Figure 2D). Between 6-8 colonies are routinely screened for each clone due to the variation of cloning efficiency (Figure 2D). Initial PCR reactions are set up with vector specific forward and reverse primers, so that a product is amplified only if the gene was inserted in the correct orientation. In the case of no product being amplified, multiple combinations of vector and/or gene specific primers can be tested. Once clones with inserts are identified, then plasmid is prepared (see above) for confirmation by DNA sequencing. - Once a colony produced a product of the expected size in colony PCR screening, the sequence of cloned inserts was confirmed by single-read paired-end Sanger dideoxy sequencing and by comparing the clone sequence to that of the reference genome. For clones longer than 1,200 bp, internal primers were designed to complete the sequence confirmation. The most common issue to arise during confirmation is the occurrence of SNPs that do not conform to the reference genome. If the SNP causes a silent mutation, or if the SNP was entered in the GRAMENE database (www.gramene.org) as a natural variant, then it was deemed acceptable for addition to the TFome collection.
- It is important to make duplicate permanent glycerol stocks of each verified clone immediately to avoid confusion in generating the TFome collection.
- DNA bands of the correct size is identified by DNA gel electrophoresis are excised and gel purified. There are many suitable kits for purification for this step. For this project we used the Wizard® SV Gel and PCR Clean-Up System. Carefully quantify the purified product prior to ligation.
- Gene synthesis of rare TF transcripts.
Chemical gene synthesis is an option for genes that prove recalcitrant to PCR amplification. This option offers the advantage that codon usage may be optimized for expression in different hosts, as is the desired downstream use of the TFome collection, and the production time is fast. However, costs are currently competitive for clones < 1 kb in length. Another disadvantage is that by not using a template derived from plant tissues, then there may be little or no direct experimental support for the existence of a particular transcript in vivo. Thus, it is recommended where possible, that RNA-Seq support be sought for a particular gene model prior to chemical synthesis.
For the maize TFome project, approximately 30% of the collection was chemically synthesized (Burdo et al., 2014). Gene synthesis was performed using the GeneArt® technology. This technology employs a codon optimization process, which is required for any complex or GC-rich sequence, and to increase expression in maize. In the case of the maize TFome, a pilot test revealed that maize optimized constructs expressed at a lower but significant level in yeast compared to yeast optimized versions. No significant difference in expression was observed between different codon optimized versions in maize protoplasts (Burdo et al., 2014). Thus, in the case of the maize TFome the chemically synthesized clones were optimized for expression in maize where most downstream experimentation is expected to be performed.
Once the fragments are synthesized they are resuspended in water and are cloned into the relevant Gateway® entry vector as described in section D above. - Storage of TFome
Given the utility of a TFome collection, careful consideration should be applied to how the TFome collection is stored and made available for long-term distribution. The maize TFome is made publicly available through the Arabidopsis Biological Resource Center (ABRC) (abrc.osu.edu). It is recommended that one or more backup TFome collections are created and stored in separate locations. Storage in a 96 well format allows for easier replication of the entire library but can make distribution of individual clones more difficult. Here we describe a method for the storage of a TFome collection in a 96 well format.
Cryogenic Storage of TFome in 96 well format.- Prepare sterilized freezing medium according to the recipe provided below which is adapted from Woo et al. (1994). This medium allows for the growth of bacterial cells and contains cryoprotectant to allow direct freezing of cultures following growth. In a laminar airflow cabinet, aliquot 1.8 ml of freezing medium containing the appropriate antibiotic into a sterile 2 ml 96 well culture dish.
- Using aseptic technique, inoculate 96 well plate with the individual bacterial stocks either from a plate or a previously made glycerol stock. When inoculating stocks, it can be helpful to have a map of the wells so as to avoid errors. Seal the plate with a breathable plate seal that is suitable for ventilating and storing bacterial/cell cultures. Place the sealed plate at 37 °C in a shaking incubator overnight with 220 rpm rotation for proper aeration.
- Since the cells are grown in freezing medium it is simply a matter of aseptically aliquoting 400 µl of the cultures into sterile 0.5 ml plates, and sealing them for storage at -80 °C. Multiple permanent plates can be made at this time. The plates may be stored at -80 °C with a plastic lid or a 7 mm sized silicone seal that can be re-autoclaved. Permanents may also be made in a 96-well format that permits automated decapping such as with the Matrix sample storage system.
- Prepare sterilized freezing medium according to the recipe provided below which is adapted from Woo et al. (1994). This medium allows for the growth of bacterial cells and contains cryoprotectant to allow direct freezing of cultures following growth. In a laminar airflow cabinet, aliquot 1.8 ml of freezing medium containing the appropriate antibiotic into a sterile 2 ml 96 well culture dish.
Representative data
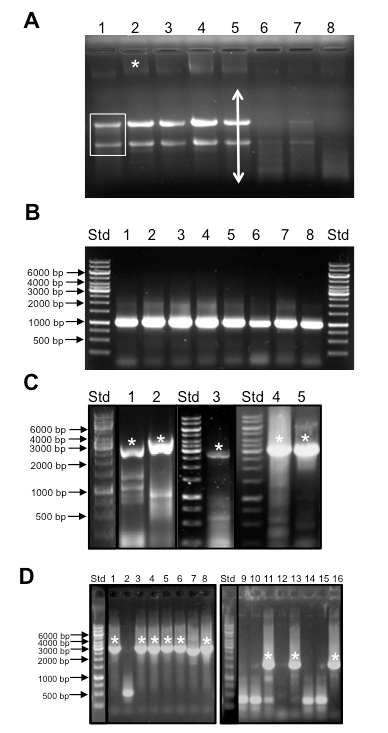
Figure 2. Representative data. Sample outcomes for plant RNA isolation and quality control, RT-PCR of long templates, and colony PCR. A. Appearance of total RNA prior to DNase treatment separated by gel electrophoresis. A non-degraded RNA sample will exhibit two major bands representing ribosomal RNAs (boxed bands in lane 1) and a background smear indicative of mRNA (arrow in lane 4). Note that diminished amounts of ribosomal RNA are seen in germinating seeds since the endosperm is not alive. A higher molecular weight band is indicative of genomic DNA that must be removed by DNase treatment. Std = Generuler™ 1 kb ladder molecular weight size standards. Samples are as follows:- 1: plant prop roots from L13 stage plant, 2: mature tassel from L13 stage plant 3: Developing seeds 2 weeks after pollination 4: entire ear 1 day after pollination, 5 seedling roots from 3 week old plant, 6, 7, and 8: germinating seeds 1, 2, and 7 days after germination. B. Amplification of GAPDH from cDNA generated from RNA isolated in panel A. A single band of 1.1 kb is indicative of the GAPDH transcript whereas a 4.3 kb band is expected if amplification occurs from genomic DNA. Std = Generuler™ 1kb ladder molecular weight size standards as in Figure 2A. cDNA samples were derived from the same samples as shown in Figure 2A. C. Amplification of TFs with long coding sequences by RT-PCR. Typically multiple bands are observed during RT-PCR from total cDNA samples. Asterisks indicate correct sized band that was excised and used for cloning. Samples and expected amplicon lengths are as follows:- 1: GRMZM2G069365, 2,127 bp, 2: GRMZM2G171600, 2,526 bp, 3: GRMZM2G028980, 2,742 bp, 4, 5: GRMZM2G160005, 3,159 bp. Std = Generuler™ 1 kb ladder molecular weight size standards as in Figure 2A. D. Sample Colony PCR reactions. Lanes with asterisks represent colonies with clones containing inserts of the expected size. High variability in the success rate of cloning different genes underscores the need for a rapid PCR based screening method. Std = Generuler™ 1 kb ladder molecular weight size standards as in Figure 2A. Lanes 1- 8: amplicons from GRMZM2G140156, expected size 2.6 kb. Lanes 9-16: amplicons from GRMZM2G009478 expected fragment size of 1.8 kb.
Notes
- The availability of a quality flcDNA library will be one of the factors that will most influence the cost of the TFome project. Amplifying rare and long transcripts from cDNA by RT-PCR can be challenging and the synthesis of long coding sequences can be prohibitively expensive.
- If no bands are observed following the initial PCR, then lowering the annealing temperature by 2 °C increments is recommended. Conversely, if multiple bands are seen then raising the annealing temperature by 2 °C increments is recommended.
- It is recommended not to allow the PCR reactions to dwell at 4 °C for any length of time at the end of the amplification cycles, as this will permit degradation of the amplicon ends and reduce cloning efficiency.
Recipes
- RNA extraction buffer I (Li and Trick, 2005) per liter per 100 ml
100 mM Tris (pH 8.0) MW 121.14 12.11g 1.211 g
150 mM LiClMW 42.39 6.34 g 0.63 g
50 mM EDTAMW 372.24 18.6 g 1.86 g
1.5 % 2-mercaptoethanol 15 ml 1.5 ml - RNA extraction buffer II stock solutions (Li and Trick, 2005) per liter per 100 ml
Stock 0.75 M sodium citrate MW 294.1 220.5 g 22 g
Stock 2 M sodium acetate (anhydrous) pH 4.0 MW 82.03 164 g 16.4 g
Note: It needs to be warmed and use glacial acetic acid to adjust to pH 4.0.
Stock 10% lauryl sarcosine MW 293.39 29.34 g 2.93 g
Note: Use a 10% stock solution, which needs to be heated to 68 °C to dissolve. - RNA extraction buffer II (working solution) (Li and Trick, 2005) per liter per 100 ml
4.2 M guanidine isothiocyanate (w/v) MW 118.16 496.27 g 49.6 g
0.5% Lauryl sarcosine Sigma (from 10% Stock) 50 ml 5 ml
1 M Sodium acetate (from 2 M stock) 500 ml 50 ml
25mM Sodium citrate (from 0.75 M stock) 33 ml 3.3 ml - Carlson lysis buffer (Carlson et al., 1991) per liter
100 mM Tris-Cl (pH 9.5) MW 121.14 12.114 g
2% CTAB (cetryl trimethyl ammonium bromide) 20.0 g
1.4 M NaCl MW 58.44 81.82 g
1% PEG 6000 or 8000 10 g
20 mM EDTA MW 372.24 (>5 M stock = 18.61 g/100 ml) 40 ml of 0.5 M stock
Need to heat to dissolve CTAB. After autoclaving the solution will appear slightly opaque (Cloudy).
Note: Add beta-mercaptoethanol to tubes just prior to use (100 µl BM per 10 ml). - Freezing medium (Woo et al., 1994) per liter
Luria-Bertani broth (LB) powder or granules 25 g
36 mM K2HPO4 MW 174.2 6.28 g
13 mM KH2PO4 MW 136.09 1.8 g
1.9 mM Na3C6H5O7∙2H2O (sodium citrate) MW 258.06 0.5 g
6.8 mM (NH4)2SO4 (ammonium sulfate) MW 132.14 0.9 g
4.4% C3H8O3 (glycerol) MW 92.09 44 ml
Bring to 1,000 ml using deionized distilled water and autoclave in large media bottles.
Separately autoclave per 100 ml
1 M MgSO4∙7 H2O (Magnesium sulfate) MW 246.475 24.6 g
Immediately prior to use, aseptically add 0.4 ml of 1M magnesium sulfate stock per liter of freezing medium and swirl to mix. The appropriate antibiotic should be also added at this time.
Acknowledgments
We appreciate the willingness of the Arabidopsis Biological Resource Center (ABRC), for accepting the TFome collection for storage, propagation and distribution. We thank Diego Mauricio Riaño-Pachón for his assistance with curation of the gene families. We thank the contributions of more than 300 University of Toledo undergraduate students who participated in the FIRE (Fostering the Integration of Research with Educational laboratory classes) program, as well as Azam Abdollahzadeh, Andrew Reed, Erik Mukundi, Evans Kataka, Narmer Fernando Galeano Vanegas, Flavia Santos, Hai-Dong Yu, Jeffrey Campbell, Tina Agarwal, Jennifer Carstens, Katja Machemer-Noonan, Kelly Scarberry, Kengo Morohashi, Kristen Belesky, Maria Tobias, Noor Zayed, Thais Andrade and Tomoe Kusayanagi and SiGuE (Success in Graduate Education, http:// www.sigue-caps.org/) fellows Miriam Mills and Gilbert Kayanja, for their outstanding contributions in team-cloning. Michael dos Santos Brito thanks FAPESP (Sao Paulo Research Foundation) for postdoctoral fellowship BEPE 2012/20486-2. Support for this project was provided by NSF IOS-1125620 to JG, AID and EG.
References
- Buitrago-Florez, F. J., Restrepo, S. and Riano-Pachon, D. M. (2014). Identification of transcription factor genes and their correlation with the high diversity of stramenopiles. PLoS One 9(11): e111841.
- Burdo, B., Gray, J., Goetting-Minesky, M. P., Wittler, B., Hunt, M., Li, T., Velliquette, D., Thomas, J., Gentzel, I., dos Santos Brito, M., Mejia-Guerra, M. K., Connolly, L. N., Qaisi, D., Li, W., Casas, M. I., Doseff, A. I. and Grotewold, E. (2014). The Maize TFome--development of a transcription factor open reading frame collection for functional genomics. Plant J 80(2): 356-366.
- Carlson, J. E., Tulsieram, L. K., Glaubitz, J. C., Luk, V. W., Kauffeldt, C. and Rutledge, R. (1991). Segregation of random amplified DNA markers in F1 progeny of conifers. Theor Appl Genet 83(2): 194-200.
- Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Heger, A., Hetherington, K., Holm, L., Mistry, J., Sonnhammer, E. L., Tate, J. and Punta, M. (2014). Pfam: the protein families database. Nucleic Acids Res 42(Database issue): D222-230.
- Gu, Z., Cavalcanti, A., Chen, F. C., Bouman, P. and Li, W. H. (2002). Extent of gene duplication in the genomes of Drosophila, nematode, and yeast. Mol Biol Evol 19(3): 256-262.
- Li, Z. and Trick, H. N. (2005). Rapid method for high-quality RNA isolation from seed endosperm containing high levels of starch. Biotechniques 38(6): 872, 874, 876.
- Perez-Rodriguez, P., Riano-Pachon, D. M., Correa, L. G., Rensing, S. A., Kersten, B. and Mueller-Roeber, B. (2010). PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res 38(Database issue): D822-827.
- Soderlund, C., Descour, A., Kudrna, D., Bomhoff, M., Boyd, L., Currie, J., Angelova, A., Collura, K., Wissotski, M., Ashley, E., Morrow, D., Fernandes, J., Walbot, V. and Yu, Y. (2009). Sequencing, mapping, and analysis of 27,455 maize full-length cDNAs. PLoS Genet 5(11): e1000740.
- Woo, S. S., Jiang, J., Gill, B. S., Paterson, A. H. and Wing, R. A. (1994). Construction and characterization of a bacterial artificial chromosome library of Sorghum bicolor. Nucleic Acids Res 22(23): 4922-4931.
- Yilmaz, A., Mejia-Guerra, M. K., Kurz, K., Liang, X., Welch, L. and Grotewold, E. (2011). AGRIS: the Arabidopsis Gene Regulatory Information Server, an update. Nucleic Acids Res 39(Database issue): D1118-1122.
- Yilmaz, A., Nishiyama, M. Y., Jr., Fuentes, B. G., Souza, G. M., Janies, D., Gray, J. and Grotewold, E. (2009). GRASSIUS: a platform for comparative regulatory genomics across the grasses. Plant Physiol 149(1): 171-180.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gray, J., Burdo, B., Goetting-Minesky, M. P., Wittler, B., Hunt, M., Li, T., Velliquette, D., Thomas, J., Agarwal, T., Key, K., Gentzel, I., Brito, M. D. S., Mejía-Guerra, M. K., Connolly, L. N., Qaisi, D., Li, W., Casas, M. I., Doseff, A. I. and Grotewold, E. (2015). Protocol for the Generation of a Transcription Factor Open Reading Frame Collection (TFome). Bio-protocol 5(15): e1547. DOI: 10.21769/BioProtoc.1547.
Category
Systems Biology > Genomics > Exome capture
Systems Biology > Genomics > Sequencing
Plant Science > Plant molecular biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.