- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Primer Extension Reactions for the PCR- based α- complementation Assay

Published: Vol 5, Iss 12, Jun 20, 2015 DOI: 10.21769/BioProtoc.1509 Views: 11330
Reviewed by: Yu ChenChang Ho LeeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The PCR- based- α- complementation assay is an effective technique to measure the fidelity of polymerases, especially RNA-dependent RNA polymerases (RDRP) and Reverse Transcriptases (RT). It has been successfully employed to determine the fidelity of the poliovirus polymerase 3D-pol (DeStefano, 2010) as well as the human immunodeficiency virus Reverse Transcriptase (HIV RT) (Achuthan et al., 2014). A major advantage of the assay is that since the PCR step is involved, even the low yield of products obtained after two rounds of low yield of RNA synthesis (for RDRP) or reverse transcription (for RT) can be measured using the assay. The assay also mimics the reverse transcription process, since both RNA- and DNA- directed RT synthesis steps are performed. We recently used this assay to show that the HIV RT, at physiologically relevant magnesium concentration, has accuracy in the same range as other reverse transcriptases (Achuthan et al., 2014). Here, we describe in detail how to prepare the inserts using the primer extension reactions. The prepared inserts are then processed further in the PCR- based- α- complementation assay.
Materials and Reagents
- pBSM13+ (Stratagene)
- T3 RNA polymerase (Roche Diagnostics, catalog number: P2083 )
- 10x transcription buffer (Roche Diagnostics, catalog number: P2083 )
- High-fidelity PvuII (PvuII-HF) (New England Biolabs, catalog number: R3151L )
- High-fidelity EcoRI (EcoRI-HF) (New England Biolabs, catalog number: R3101L )
- 10x CutSmart buffer (New England Biolabs, catalog number: R3101L)
- 6x gel loading dye (New England Biolabs, catalog number: R3101L)
- NdeI (New England Biolabs, catalog number: R0111L )
- T4 polynucleotide kinase (PNK) (New England Biolabs, catalog number: M0201L )
- 10x T4 polynucleotide kinase buffer (New England Biolabs, catalog Number: B0201S )
- RNasin (RNase inhibitor) (New England Biolabs, catalog number: M0307L )
- RNase (DNase-free) (Roche Diagnostics, catalog number: 11119915001 )
- RNase-free DNase I (Affymetrix, catalog number: 784111000 )
- Pfu DNA polymerase (Agilent Technologies, catalog number: 600353 )
- 10x Pfu buffer (Agilent Technologies, catalog number: 600353)
- Ribonucleoside triphosphate set (Roche Diagnostics, catalog number: 11277057001 )
- Deoxynucleoside triphosphate (dNTP) (Roche Diagnostics, catalog number: 11969064001 )
- Gamma [γ-32P] ATP (PerkinElmer, catalog number: Blu502A001MC )
- G-25 Macro spin columns (best suited for volumes of 75-150 μl) (Harvard Apparatus, catalog number: 74-3901 )
- RNeasy RNA purification kit (QIAGEN, catalog number: 74104 )
- Phenol: Chloroform: Isoamyl alcohol (25:24:1) (Amresco, catalog number: K169-400ML )
- Ethanol (VWR Lifesciences, catalog number: EM1.00967.4003 )
- 3M Sodium Acetate (Amresco, catalog number: E521-100ML )
- Isopropyl alcohol (J.T.Baker®, catalog number: 9037-03 )
- 40% Acrylamide-Bisacrylamide (19:1) solution (VWR International, catalog number: JT4968-0 )
- 40% Acrylamide-Bisacrylamide (29:1) solution (VWR International, catalog number: JT4968-0 )
- Urea (VWR International, catalog number: 97061-926 )
- Ammonium Persulfate (VWR International, catalog number: 97064-594 )
- HIV Reverse Transcriptase, [purified as described in Hou et al. (2004)]
- Milli-Q quality [RNase, DNase free water (dH2O)]
- DNA oligonucleotides were obtained from Integrated DNA Technologies
- Extension reaction buffer (see Recipes)
- Elution buffer (see Recipes)
- 2x SDS loading buffer (see Recipes)
Equipment
- Eppendorf tubes
- Micropipette
- Petri plates
- Table top centrifuge
- Incubator
- Gel apparatus
Procedure
- Primer labelling
- All the primers should be first radiolabelled in 50 µl of 1x PNK buffer along with 50 pico moles of each primer, 10 μl of [γ-32P] ATP and 5 units of polynucleotide kinase (PNK). The reaction mixture was incubated for 30 min at 37 °C and the PNK was heat inactivated for 15 min at 65 °C.
- G-25 spin columns were incubated with 500 µl dH2O for 15 min to equilibrate the column and the excess water was removed by spinning the columns at a table top centrifuge at 5,000 rpm for 4 min.
- After heat inactivation, the excess [γ-32P] ATP was removed from the reaction mixture by loading it onto an equilibrated column and spinning at 5,000 rpm for 4 min.
- All the primers should be first radiolabelled in 50 µl of 1x PNK buffer along with 50 pico moles of each primer, 10 μl of [γ-32P] ATP and 5 units of polynucleotide kinase (PNK). The reaction mixture was incubated for 30 min at 37 °C and the PNK was heat inactivated for 15 min at 65 °C.
- Preparation of RNA for fidelity assay
- The transcript used as a template for the fidelity assay was derived from the plasmid pBSΔPvuII1146, which was prepared as described in DeStefano et al. (1998).
- For preparation of the RNA, 10 μg of the plasmid was cleaved with 50 units of the enzyme NdeI in the NEB buffer 4 for 3 h at 37 °C. The cleaved plasmid was then extracted with phenol chloroform extraction and recovered by ethanol precipitation as described below.
- After cleavage, run-off transcription was performed in 100 µl of the transcription buffer along with 2 µg of the linearized plasmid, 5 µl of 100 mM DTT, 10 µl of 5 mM ribonucleotides, 2 µl of RNase inhibitor, and 40 units of T3 RNA polymerase for 3 h at 37 °C.
- 10 units of DNase I was added to the reaction mixture and the reaction was incubated for 10 min to digest the remaining DNA.
- The RNA was then purified using the QIAGEN RNeasy kit, as per the manufacturer’s instructions, and quantified using the spectrophotometer.
- The transcript used as a template for the fidelity assay was derived from the plasmid pBSΔPvuII1146, which was prepared as described in DeStefano et al. (1998).
- RNA-directed DNA synthesis
- The ~760 nt RNA template, prepared using the method described above, was hybridized to a radiolabeled 25-nt DNA primer (5ʹ-GCGGGCCTCTTCGCTATTACGCCAG-3ʹ).
- For hybridization, 50 nM of the primer was added to 25 nM of the template (2:1 ratio of primer: template) in 48 μl of the extension reaction buffer along with 6 mM MgCl2 and 100 μM dNTPs. The mixture was heated at 65 °C for 5 min and then slowly cooled to room temperature. The total reaction volume was 50 μl.
- The primer- template was incubated at 37 °C for 3 min. 2 μl of 5 μM HIV RT was added to initiate the extension reaction and the incubation was continued for 30 min. Full extension of the primer should yield a 199 nucleotide (nt) DNA product (Figure 1).
Note: The primer used here was diluted 10-fold with unlabeled primer so that the extension product from this round will have less specific radioactivity than the product from the next round of synthesis (Figure 2).

Figure 1. Schematic illustration of the assay. RNAs are indicated by broken lines and DNAs by solid lines. Primers have arrowheads at the 3ʹ end. The ~760-nucleotide template RNA used as the initial template for HIV RT RNA-directed DNA synthesis is shown at the top with the 3ʹ and 5ʹ ends indicated. The positions of PvuII and EcoRI restriction sites are indicated for reference to the vector. Numbering is based on that for the plasmid pBSM13+.
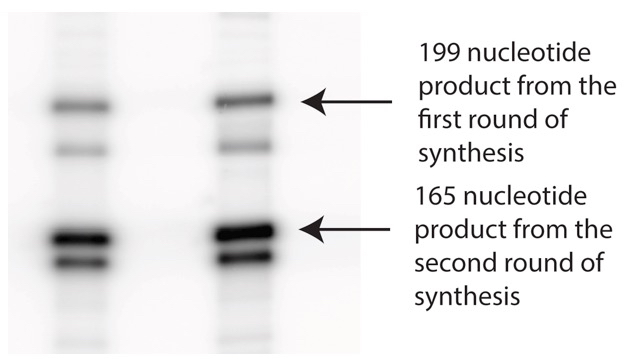
Figure 2. Representative Data after two rounds of synthesis. Products from the DNA-directed synthesis appear darker in the gel than the products from the first round because the primers used for this step had higher specific activity.
- After 30 min, 1 μl of RNase was added to digest the remaining RNA and the sample was heated to 65 °C for 5 min to deactivate the RT.
- The DNA product was then recovered by standard phenol chloroform extraction. Equal volume (50 μl) of phenol: chloroform: isoamyl alcohol mixture (25:24:1) was added to the reaction. The reaction was mixed well and then spun in the microcentrifuge at 18,000 rpm for 3 min. The aqueous layer from the top was removed, transferred to a new tube and equal volume (~50 μl) of chloroform was added. The mixture was spun again at 18,000 rpm for 3 min in the microcentrifuge and the aqueous layer was removed carefully and moved to a new tube.
- DNA product was precipitated using standard ethanol precipitation. Two volumes of 100% ethanol (100 μl) and one-tenth volume (5 μl) of 3M sodium acetate were added and the reaction was stored at -20 °C for two hours.
- After two hours, the reaction was spun for 20 min at 18,000 rpm. The supernatant was discarded carefully. The pellet was washed with 500 µl of 70% ethanol and then dried.
- The pellet was resuspended in 2x loading buffer and the products were separated on a 6% denaturing 7 M urea-polyacrylamide gels.
- 199 nt DNA product was cut out from the gel and the gel pieces was crushed and re-suspended in 500 μl elution buffer. After overnight elution at 4 °C, the material was filtered through 0.45 μM syringe filter and the DNA product was recovered using phenol chloroform extraction method described above.
- The recovered product was resuspended in elution buffer, quantified using a spectrophotometer, and used as a template for the DNA directed DNA synthesis step.
- The ~760 nt RNA template, prepared using the method described above, was hybridized to a radiolabeled 25-nt DNA primer (5ʹ-GCGGGCCTCTTCGCTATTACGCCAG-3ʹ).
- DNA directed DNA synthesis
- The recovered DNA product from RNA directed synthesis was hybridized as described above to another 20-nt radiolabeled DNA primer (5ʹ-AGGATCCCCGGGTACCGAGC-3ʹ) with at least 10-fold-greater specific activity than the primer used for round 1 (see above).
- A second round of DNA synthesis was performed as described above except that the reaction volume was 25 μl. Conditions for extension were maintained identical in the RNA and DNA template extension reactions.
- Reactions were terminated with 25 µl of 2x loading buffer, and products were gel purified on an 8% denaturing 7 M urea-polyacrylamide gels.
Note: The gel was run far enough to efficiently separate the 162-nt full extension product of round 2 from the 199-nt template (Figure 2).
- The 162 nt product was located and cut out from the gel. The product was recovered as described above and resuspended in 100 μl of elution buffer.
- The recovered DNA product from RNA directed synthesis was hybridized as described above to another 20-nt radiolabeled DNA primer (5ʹ-AGGATCCCCGGGTACCGAGC-3ʹ) with at least 10-fold-greater specific activity than the primer used for round 1 (see above).
- PCR reactions
- The round 2 DNA produced in the above-described step was amplified by PCR using the following primers: 5ʹ-GCGGGCCTCTTCGCTATTACGCCAG-3ʹ and 5ʹ-AGGATCCCCGGGTACCGAGC-3ʹ.
- The first primer is the one used for the RNA directed DNA synthesis and the second primer overlaps a region of the EcoRI site on the plasmid from which the RNA was originally transcribed.
- PCR reactions were performed in the purchased 1x Pfu buffer along with 200 μM dNTPs, 50 pico moles of each primer, and 5 units of Pfu polymerase. 5 μl of the product from the DNA directed DNA synthesis was used as a template. The total reaction volume was 50 μl.
Note: Initial denaturation step at 95 °C (5 min) followed by twenty-five PCR cycles at 94 °C (1 min), 50 °C (1 min), 72 °C (1 min), and finally a 5 min incubation at 72 °C were performed.
- The aqueous phase from the PCR reactions was phenol-chloroform extracted and recovered by ethanol precipitation as described above.
- PCR products were digested with 30 units each of EcoRI and PvuII in a total of 75 μl CutSmart buffer for 1 h at 37 °C.
- 15 μl of 6x gel loading dye was added to each reaction and samples were electrophoresed on a 12% non-denaturing polyacrylamide gel.
- The DNA was located by UV shadowing, recovered by overnight elution as described above, and spectrophotometry was used to quantify the recovered DNA.
After preparing the inserts using the primer extension reactions and the PCR reactions described above, the plasmid vector was then prepared as described in DeStefano (2010). The inserts and the plasmid vector were ligated and the ligated plasmid was transformed into GC-5 E.coli cells as described in DeStefano (2010). Blue, faint blue, and white colonies were then counted and the colony mutation frequency of RT was determined using the number of each colonies.
Note: All insertion or deletion frameshift mutations and most of the substitution mutations introduced by RT during either the RNA-directed or the DNA-directed synthesis steps will result in a faint blue or a white colony. The ratio of the number of faint blue/white colonies to the total number of colonies will give the colony mutation frequency (CMF). For example: In an experiment conducted at 6 mM Mg2+, 32 faint blue/white colonies were obtained along with 3605 blue colonies giving a CMF value of 8.8 x 10-3.
- The round 2 DNA produced in the above-described step was amplified by PCR using the following primers: 5ʹ-GCGGGCCTCTTCGCTATTACGCCAG-3ʹ and 5ʹ-AGGATCCCCGGGTACCGAGC-3ʹ.
Recipes
- Extension reaction buffer (50 ml)
1 M Tris HCl (pH 8)
25 ml
3 M KCl
13.3 ml
1 M DTT
1ml
RNase free water
10.7 ml
- Elution buffer (50 ml)
1 M Tris HCl (pH 7.5)
500 μl
0.5 M EDTA
100 μl
RNase free water
49.4 ml
- 2x SDS loading buffer (10 ml)
50 mM Tris HCl pH 6.8
500 μl
100 mM DTT
1 ml
2% SDS
2 ml
0.05% Bromophenol blue
500 μl
10% glycerol
1 ml
RNase free water
5 ml
References
- Achuthan, V., Keith, B. J., Connolly, B. A. and DeStefano, J. J. (2014). Human immunodeficiency virus reverse transcriptase displays dramatically higher fidelity under physiological magnesium conditions in vitro. J Virol 88(15): 8514-8527.
- DeStefano, J. J. (2010). Effect of reaction conditions and 3AB on the mutation rate of poliovirus RNA-dependent RNA polymerase in a alpha-complementation assay. Virus Res 147(1): 53-59.
- DeStefano, J., Ghosh, J., Prasad, B. and Raja, A. (1998). High fidelity of internal strand transfer catalyzed by human immunodeficiency virus reverse transcriptase. J Biol Chem 273(3): 1483-1489.
- Hou, E. W., Prasad, R., Beard, W. A. and Wilson, S. H. (2004). High-level expression and purification of untagged and histidine-tagged HIV-1 reverse transcriptase. Protein Expr Purif 34(1): 75-86.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Achuthan, V. and DeStefano, J. J. (2015). Primer Extension Reactions for the PCR- based α- complementation Assay. Bio-protocol 5(12): e1509. DOI: 10.21769/BioProtoc.1509.
Category
Microbiology > Microbial biochemistry > Protein > Activity
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.