- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative Image Analysis of Membrane Microdomains Labelled by Fluorescently Tagged Proteins in Arabidopsis thaliana and Nicotiana benthamiana


Published: Vol 5, Iss 11, Jun 5, 2015 DOI: 10.21769/BioProtoc.1497 Views: 10108
Reviewed by: Arsalan DaudiRenate WeizbauerAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
We have recently characterized co-existing membrane microdomains that are labeled by different proteins in living plant cells (Jarsch et al., 2014). For this approach we first created a digital fingerprint for each of the twenty marker proteins using quantitative image analysis. Here we recorded parameters such as domain size, density and shape based on image segmentation. We found highly reproducible patterns of any of the proteins over a large number of biological replicates. Furthermore we exclusively acquired images from lowly expressing cells and chose our imaging conditions in a way that resulted in images where no pixel was saturated.
This protocol describes in detail the methods that have been used to analyze quantitative differences in localization of members of the remorin protein family in membrane microdomains of Arabidopsis thaliana and Nicotiana benthamiana (Jarsch et al., 2014). The proteins were either individually or pairwise expressed as fluorophore fusions in the respective plant. Image acquisition was performed using standard Confocal Laser Scanning Microscopy (CLSM) and image analysis was performed using ImageJ.
[Introduction] Since confocal laser-scanning or other state-of-the-art fluorescence microscopes are nowadays often regarded as standard equipment a modern research institution should have, the amount of published cell biological data has massively increased over the last years. This certainly also correlates with the availability of an increasing number of fluorophores and corresponding expression vectors that have made it comparably easy to generate large numbers of tagged proteins. One main concern about showing microscopy images in publications is the subjectivity they have been selected with. In addition, and certainly very unfortunate in several cases, the scientific community as well as reviewers of manuscripts have requested ‘no background-high fluorescence’ images from the authors. As a consequence researchers often started selecting the images based on aesthetic aspects rather than showing the most representative ones. Furthermore the majority of images are based on strong over-expression of proteins. Therefore quantitative image analysis has become an absolute requirement in order to make robust statements on cell biological observations and the frequency with which they have been observed. However, this does not only require gaining novel skills but also high numbers of biological repetitions in a standardized way. Furthermore, it should be the ultimate goal to work under conditions where the protein of interest is expressed at native levels. While this may have to be overcome for lowly abundant proteins, researchers should nevertheless aim for similar levels and may thus accept more background noise in the images.
It should be noted that all parameters and protocol specifications provided within this protocol have been optimized for the expression we used in a current study (Jarsch et al., 2014). Most likely they have to be adapted for any analyses in different laboratories.
Materials and Reagents
- 4-5 weeks old Nicotiana benthamiana (N. benthamiana) plants (soil grown)
- 4-5 weeks individually potted stable transgenic Arabidopsis thaliana (A. thaliana) lines expressing AvrPto under control of a dexamethasone (DEX)-inducible promoter as described previously (Hauck et al., 2003; Tsuda et al., 2012) (lines available upon request from the authors of the original publication) (soil grown)
- Agrobacterium strains
- GV3101 C58 mp90RK for constructs in pAM-PAT:35S and pH7YGW2
- Agl1 for constructs in pUbi and pGWB1-based vectors
- GV3101 C58 mp90RK for constructs in pAM-PAT:35S and pH7YGW2
- MgCl2 (Carl Roth, catalog number: 2189 )
- MES KOH (pH 5.6) (Carl Roth, catalog number: 4256.2 )
- Acetosyringone (Sigma-Aldrich, catalog number: D134406 )
- Dexamethasone (Sigma-Aldrich, catalog number: D4902 )
- EtOH
- Silwett L-77 (Leu+Gygax AG, catalog number: CH-SL7-033-01 )
- Infiltration solution for Agrobacterium tumefaciens-mediated transient transformation of N. benthamiana or A. thaliana (see Recipes)
- DEX-solution for pre-treatment of AvrPto-DEX inducible A. thaliana (see Recipes)
Equipment
- Table-top centrifuge for 2 ml tubes (Eppendorf, model: 5424 )
- Spectrometer (Pharmacia Biotech (now: GE Healthcare, Ultrospec 3000 pro)
- 1 ml syringes (Braun catalog number: 9161406V )
- 2 ml reaction tubes for centrifugation (SARSTEDT AG, catalog number: 72.695.500 )
- 50 ml spray flask (Carl Roth, Karlsruhe, catalog number: EP66 )
- 4 mm biopsy punch or similar tool (cork borer) to excise leaf discs (recommended: Produkte für Medizintechnik (pfm), 4 mm, catalog number: 49401 )
- Microscope cover glasses (Carl Roth, 24 x 60 mm, #1,5 (170 micron), catalog number: H878 )
- Microscope slides (Langenbrick, 76 x 26 x 1 mm, catalog number: 03-0010/90 )
- Confocal microscope (Leica Microsystems, model: SP5 )
- Leica DFC350FX digital camera
- Objectives used for this experiment: HC PL APO 20x/0.70 ImmCorr CS and HCX PL APO 63x/1.20 W CORR CS)
- Excitation using an argon laser (100 mW, Lasos LGK 7872 ML05 SP5) [for YFP: 514 nm (excitation), 525-600 nm (emission); for CFP: 456 nm (excitation); 475-620 nm (emission)]
Software
- ImageJ (Plug-in supplemented version: Fiji)
- Intensity Correlation Analysis Bundle from the Wright Cell Imaging Facility (WCIF Image) (Li et al., 2004)
Procedure
A. tumefaciens-mediated transient transformation of N. benthamiana
- Culture bacterial strains carrying the constructs of choice in 5 ml liquid LB under appropriate antibiotic selection overnight (ON) at 28 °C.
- Harvest bacteria by centrifugation in 2 ml reaction tubes at 6,500 x g and resuspend in infiltration solution. For pUbi-YFP constructs use a final OD600 of 0.01 and for pAM-PAT-35S-CFP/YFP constructs use a final OD600 of 0.2-0.4.
- To improve expression, add an A. tumefaciens Agl1 strain containing a construct mediating expression of the viral silencing inhibitor P19 (Voinnet et al., 2003) to each sample at a final OD600 of 0.1. Excess presence of the same type of RNA may cause RNA degradation by the plant as part of the immune response to a putative viral infection. P19 helps to reduce such RNA degradation.
- Incubate the solution for 2 h in the dark at room temperature (RT).
- Subsequently syringe-infiltrate the youngest fully expanded leaves with the individual samples using a 1 ml syringe (as demonstrated in Video 1). The infiltrated area will be clearly visible. Use sufficient infiltration solution to cover sufficient leave area for later imaging (usually 100-300 µl).
 Video 1. Infiltration of Agrobacterium tumefaciens into Nicotiana benthamiana leaves
Video 1. Infiltration of Agrobacterium tumefaciens into Nicotiana benthamiana leaves - Continue plant growth for 2 days prior to imaging. Water plants moderately. The time between infiltration and imaging strongly depends on the type of protein (e.g. solubility of transmembrane), the used promoters and the growth conditions of the N. benthamiana plants.
A. tumefaciens-mediated transient transformation of A. thaliana
- 24 h prior to infiltration, spray five to six weeks old plants grown under short-day conditions with the 2 μM DEX solution containing 0.04% Silwett-77. Spray under fume hood to prevent inhalation of Silwet-77. Turn plants multiple times and ensure leaves are completely covered by a thin film of liquid until drip-of. In case of drops forming on the leave surface with the liquid not spreading to make a film, slightly increase the Silwett-77 concentration.
- Prepare the infiltration solution containing A. tumefaciens strains of your choice as described for transformation of N. benthamiana above.
- Subsequently syringe-infiltrate the youngest fully expanded leaves with the individual samples using a 1 ml syringe (see Video 1 for the corresponding infiltration procedure in N. benthamiana). The infiltrated area will be clearly visible. Use sufficient infiltration solution to cover sufficient leave area for later imaging (usually 100-300 µl).
- Grow plants for 2 days for imaging, water moderately.
- Sample preparation for image acquisition
- For microscopic analysis, cut leaf discs from infiltrated areas using biopsy punches or cork borers.
- Prepare cover glasses with drops of water.
- Mount lower side of the leaf on cover slips on top of water drops, make sure there is no air between leaf tissue and glass.
- Add microscope slides and image directly.
- For microscopic analysis, cut leaf discs from infiltrated areas using biopsy punches or cork borers.
- Image acquisition
- In transiently expressing systems, choose lowly expressing cells. To be able to differentiate between autofluorescence and real fluorophore signal, choose a non-infiltrated cell area and adjust imaging settings in order to eliminate background. Use these setting to search for cells displaying non-saturating intensities. In an 8-bit greyscale image these values should ideally not exceed 100 (out of 255 possible grey levels).
- For stably expressing system, use lines, if possible, expressing your protein of interest under control of its native promoter (ideally in a mutant background). Select lines where expression of the transgene is similar to levels of the endogenous transcript (e.g. by quantitative Real Time PCR) or compare protein levels if an antibody against the endogenous protein is available.
- For single pictures of PM surfaces, use 2 line averages per frame.
- For co-localization experiments (especially when using fluorophores with partially overlapping excitation/emission spectra), use the microscope in the sequential scanning mode to avoid ‘bleed-through’ between the channels.
- Export images as .tif files for further processing or use original microscope files if readable by the processing software.
- To be able to compare images, make sure settings are identical between different takes (e.g. laser intensity, gain, offset, scan speed, etc.…)
- In transiently expressing systems, choose lowly expressing cells. To be able to differentiate between autofluorescence and real fluorophore signal, choose a non-infiltrated cell area and adjust imaging settings in order to eliminate background. Use these setting to search for cells displaying non-saturating intensities. In an 8-bit greyscale image these values should ideally not exceed 100 (out of 255 possible grey levels).
- Image processing
Quantitative analysis of subcellular single protein localization: The analysis includes the creation of a mask, which will be overlaid with the original image for measurements.- Open individual pictures of single infiltrations in Fiji.
- If necessary, change properties according to your image setting (adjust pixel and voxel size).
- Change Image → Type to 8-bit.
- Image → Duplicate image and continue working on the duplicated image.
- Run Process → Filters → Mean filter of 2 pixels.
- Subject to Process → Subtract background choosing a rolling ball radius of 20 pixels (be aware that background subtraction is critical and should be carefully evaluated prior to application. E. g. measure the size of the objects you are interested in and make sure the radius you chose is significantly bigger than your biggest object!).
- Image → Adjust → Threshold to segment the image in foreground and background, try this for a number of different images and then decide on a default setting to process all images you want to compare.
- Process → Binary → Convert to mask.
- Process → Binary → Create a binary image.
- In case of overlapping regions of interest, apply Plugins → Watershed.
- Choose Analyze → Set measurements and define which parameters you would like to analyze, tick “add to Roi manager”.
- Analyze particles (the output will be a table including all parameters previously defined in the “Set measurements” menu).
- Select the original image, click “Show all” and “Measure” in the ROI manager.
- The original image will show all regions selected by the mask for measurements, the table will now include a second set of measurements for the original image.
- To perform statistical analysis on the results we recommend R using ANOVA and Tukey’s honestly significant difference.
- Open individual pictures of single infiltrations in Fiji.
Representative data
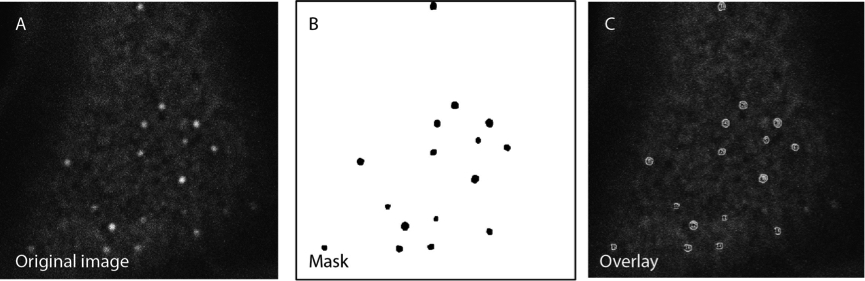
Figure 1. Creation of a binary image to segment the picture. The mask B is used as an overlay onto the original image A to carry out the desired analysis on a non-processed picture.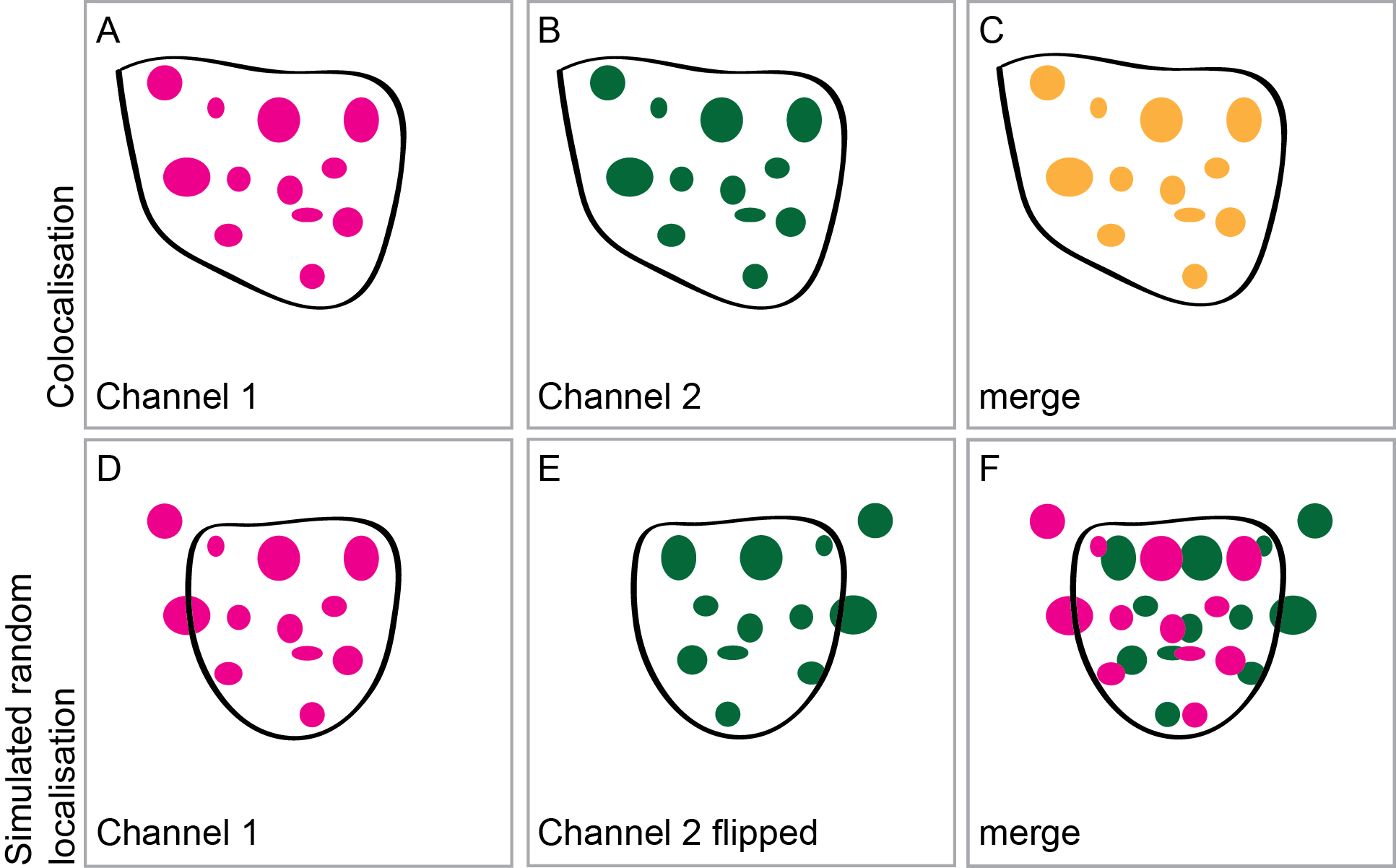
Figure 2. Intensity correlation analysis and simulation of a random distribution. Two fusion proteins A, B were expressed to assess intensity correlation. The merged imaged C should be used to confirm that the ROI was chosen appropriately, including intensities in both channels and excluding regions of the image without signal or containing out-of-focus intensities. To simulate a random distribution of the two proteins D-F one of the two images is flipped either horizontally or vertically E and again the intensity correlation analysis is carried out using a ROI chosen to exclude parts of the picture which are not suitable.
Notes
- To optimize the segregation process, alter the rolling ball radius for the background subtraction or the threshold settings. In case of salt and pepper noise, addition of “Erode” and “Dilate” steps on the binary image might be necessary.
The steps for the creation of the mask can be automatized using a Macro (see example).
Example Macro:
run("Mean...", "radius=2");
run("Subtract Background...", "rolling=20");
setAutoThreshold("Default");
//run("Threshold...");
setThreshold(0, 22);
setOption("BlackBackground", false);
run("Convert to Mask");
run("Make Binary");
run("Erode");
run("Erode");
run("Dilate");
run("Dilate");
run("Watershed");
run("Save");
run("Close"); - Intensity correlation analysis to assess of pairwise expressed proteins in a quantitative manner
- Open single images in ImageJ.
- Subject to Process → Filters → Mean Blur Filter of 2 pixels.
- Apply Process → Subtract background with a rolling ball radius of 20 pixels.
- Draw a region of interest around the area containing the intensities you are interested in, if necessary (exclude regions without signal, auto-fluorescence of out-of focus fluorescence).
- Open the Intensity correlation Plugin from WCIF ImageJ (link for download: http://www.uhnresearch.ca/facilities/wcif/fdownload.html).
- Choose “use ROI” if applicable.
- Tick options you are interested in, run.
- The Pearson correlation coefficient (Rr) (Manders et al., 1992) and the Manders overlap coefficient (R2) (Manders et al., 1993) as well as pixel ratios (Ch1: Ch2), Mander’s Colocalization coefficients for channel 1 (M1) and channel 2 (M2), the number of pixel pairs that have a positive PDM value (N+ve), the number of pixels pairs in the images that where at least one of the pixel pairs is above zero (Ntotal) and the Intensity Correlation Quotient (ICQ) will be displayed in a separate window showing a table that can be saved as a excel file.
- To define significantly positive or negative covariance of intensities, a simulated random distribution should be addressed by flipping or rotating one of the images. The results of at least 10 repetitions can be statistically analyzed using standard student TTest.
- Open single images in ImageJ.
Recipes
- Infiltration solution for A. tumefaciens mediated transient transformation of N. benthamiana or A. thaliana
10 mM MgCl2
10 mM MES KOH (pH 5.6)
150 μM acetosyringone
Agrobacteria in appropriate OD - DEX-solution for pre-treatment of AvrPto-DEX inducible A. thaliana
2 μM dexamethasone
1% EtOH
0.04% Silwett L-77
Acknowledgments
This work was kindly supported by the Sonderforschungsbereich SFB924 funded by the Deutsche Forschungsgemeinschaft (DFG). The original work was published in Jarsch et al. (2014).
References
- Hauck, P., Thilmony, R. and He, S. Y. (2003). A Pseudomonas syringae type III effector suppresses cell wall-based extracellular defense in susceptible Arabidopsis plants. Proc Natl Acad Sci U S A 100(14): 8577-8582.
- Jarsch, I. K., Konrad, S. S., Stratil, T. F., Urbanus, S. L., Szymanski, W., Braun, P., Braun, K. H. and Ott, T. (2014). Plasma membranes are subcompartmentalized into a plethora of coexisting and diverse microdomains in Arabidopsis and Nicotiana benthamiana. Plant Cell 26(4): 1698-1711.
- Li, Q., Lau, A., Morris, T. J., Guo, L., Fordyce, C. B. and Stanley, E. F. (2004). A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci 24(16): 4070-4081.
- Manders, E. M., Verbeek, F. J., and Aten, J. A. (1993). Measurement of co-localization of objects in dual-colour confocal images. J Microscopy 169: 375-382.
- Manders, E. M., Stap, J., Brakenhoff, G. J., van Driel, R. and Aten, J. A. (1992). Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103 (Pt 3): 857-862.
- Tsuda, K., Qi, Y., Nguyen le, V., Bethke, G., Tsuda, Y., Glazebrook, J. and Katagiri, F. (2012). An efficient Agrobacterium-mediated transient transformation of Arabidopsis. Plant J 69(4): 713-719.
- Voinnet, O., Rivas, S., Mestre, P. and Baulcombe, D. (2003). An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J 33(5): 949-956.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jarsch, I. K. and Ott, T. (2015). Quantitative Image Analysis of Membrane Microdomains Labelled by Fluorescently Tagged Proteins in Arabidopsis thaliana and Nicotiana benthamiana. Bio-protocol 5(11): e1497. DOI: 10.21769/BioProtoc.1497.
Category
Plant Science > Plant cell biology > Cell imaging
Biochemistry > Protein > Fluorescence
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.