- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of Total and 2-LTR (Long terminal repeat) HIV DNA, HIV RNA and Herpesvirus DNA in PBMCs


Published: Vol 5, Iss 11, Jun 5, 2015 DOI: 10.21769/BioProtoc.1492 Views: 15220
Reviewed by: Vaibhav B ShahVamseedhar RayaproluAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Almost all individuals infected with human immunodeficiency virus (HIV) are also infected with cytomegalovirus (CMV) and Epstein Barr virus (EBV). The aims of our studies have included characterizing and measuring the latent HIV reservoir and understanding the association between asymptomatic replication of CMV (and other herpesvirus, including EBV) and this HIV reservoir (Gianella et al., 2014). This protocol was designed to simultaneously co-extract DNA and RNA from the same peripheral blood mononuclear cell (PBMC) aliquot and quantify HIV, CMV and EBV DNA, as well as HIV RNA using droplet digital PCR (ddPCR).
For collection and processing of male genital secretions and quantification of HIV RNA and DNA from seven human herpesviruses from seminal plasma, refer to protocol “Quantification of HIV RNA and Human Herpesvirus DNA in Seminal Plasma” (Vargas-Meneses et al., 2015).
Materials and Reagents
- Cell processing and flow cytometry
- 1x Dulbecco’s phosphate buffered saline (DPBS) (Corning, catalog number: 21-031-CV )
- RPMI 1640 medium (Life Technologies, Gibco®, catalog number: 11875-093 )
- GemCell U.S. origin fetal bovine serum (FBS) (Gemini Bio Products, catalog number: 100-500 )
- 37% formaldehyde solution (Sigma-Aldrich, catalog number: F8775-500ML )
- Sodium azide Reagents Plus (NaN3) (Sigma-Aldrich, catalog number: S2002-25G )
- Antibodies
- AllPrep DNA/RNA Mini Kit (QIAGEN, catalog number: 80204 )
- Staining flow cytometry buffer (staining buffer) (see Recipes)
- 1% formaldehyde (1% FA) buffer (see Recipes)
- RLT plus buffer + β-ME (see Recipes) (included in the AllPrep DNA/RNA Mini Kit)
- 70% ethanol buffer (see Recipes)
- 10 mM Tris elution buffer (see Recipes)
- 1x Dulbecco’s phosphate buffered saline (DPBS) (Corning, catalog number: 21-031-CV )
- DNA/RNA co-extraction and DNA precipitation
- RNase-free DNase set (QIAGEN, catalog number: 79254 )
- AllPrep DNA/RNA Mini Kit (QIAGEN, catalog number: 80204)
- Molecular biology grade sterile purified water (RNase, DNase, proteinase free)
- Ethyl alcohol pure (200 Proof molecular biology grade) (Sigma-Aldrich, catalog number: E7023 )
- 3 M NaOAc (sodium acetate) (Affymetrix, catalog number: 75897 )
- UltraPure glycogen (Life Technologies, catalog number: 10814010 ) (concentration: 20 µg/µl)
- 2-Mercaptoethanol (β-ME) (Life Technologies, Gibco®, catalog number: 21985-023 )
- Tris hydrochloride (1 M solution pH 8.0 molecular biology grade) (Thermo Fisher Scientific, catalog number: BP1758-500 )
- RNase-free DNase set (see Recipes)
- RNase-free DNase set (QIAGEN, catalog number: 79254 )
- ddPCR
- Ban-II restriction enzyme (New England Biolabs, catalog number: R0119L )
- Primers and probes (Table 1, IDT.) (Final PCR concentration: 900 nM primer, 250 nM probe)
- iScript advanced cDNA synthesis kit (Bio-Rad Laboratories, catalog number: 172-5038 )
- ddPCR supermix for probes (no dUTP) (Bio-Rad Laboratories, catalog number: 1863024 )
- Droplet generator cartridges and gaskets (Bio-Rad Laboratories, catalog number: 1864007 )
- Droplet generator oil (Bio-Rad Laboratories, catalog number: 1863005 )
- Droplet reader oil (Bio-Rad Laboratories, catalog number: 1863004 )
- 2x buffer control (Bio-Rad Laboratories, catalog number: 1863052 )
- Pierceable foil heat seal (Bio-Rad Laboratories, catalog number: 1814040 )
- Multichannel reagent trough
- Molecular biology grade sterile purified water (RNase, DNase, proteinase free)
- Ban-II restriction enzyme (New England Biolabs, catalog number: R0119L )
Equipment
- Cell processing and flow cytometry
- 96-well plate V-bottom (VWR International, catalog number: 12-565-460 )
- Water bath at 37 °C
- BD Accuri (with plate loader) (BD, catalog number: 653118 ) or equivalent flow cytometer
- SorvallTM RC4 general purpose floor model centrifuge (Thermo Fisher Scientific, catalog number: 75004481 ) or equivalent
- 96-well plate V-bottom (VWR International, catalog number: 12-565-460 )
- DNA/RNA co-extraction and DNA precipitation
- Collection tubes (2 ml) (QIAGEN, catalog number: 19201 )
- BD 3 ml Leur-LokTM Syringe with detachable BD 23G 1½ PrecisionGlideTM needle (BD, catalog number: 309589 )
- BD 20G 1½ PrecisionGlideTM needle (BD, catalog number: 305176 )
- Sterile 1.5 ml microcentrifuge Eppendorf tubes
- Sterile 1.5 ml Micro tubes with screw cap
- Pasteur pipette (transfer pipette)
- Vortex
- Heating block
- Table centrifuge
- NanoDrop 2000 Spectrophotometer
- Collection tubes (2 ml) (QIAGEN, catalog number: 19201 )
- ddPCR
- 96-Well PCR clean plate
- Strip caps
- Plate sealing adhesive film
- 96-Well semi-skirted PCR plate (Eppendorf, catalog number: 951020362 )
- Low retention pipette tips (200 µl and 20 µl)
- 1.5 ml screw-top tubes
- Biorad droplet generator and reader, QX200 droplet digital PCR system (Bio-Rad Laboratories, catalog number: 186-4001 )
- PX1 PCR plate sealer (Bio-Rad Laboratories, catalog number: 181-4000 )
- Thermocycler
- Special multichannel pipette for ddPCR droplet transfer (P-50) (e.g. Rainin, catalog number: L8-50XLS+ )
- Centrifuge to spin plates
- Spectrophotometry (Nanodrop)
- 96-Well PCR clean plate
Procedure
- RNA and DNA co-Extraction (Qiagen’s AllPrep DNA/RNA mini kit)
- For each sample to be extracted prepare one 1.5 ml screw-cap tube and add 300 µl of 1x DPBS.
- If flow cytometry staining is to be performed, prepare a V-bottom 96 well-plate and add 150 µl of RPMI containing 20% FBS to each sample well.
Note: To normalize the data, percentage of CD4+ CD3+ T-cells will be assessed for each sample. If these data are already available, skip steps A2 and A4. - Quickly thaw 5 million peripheral blood mononuclear cells (PBMCs) aliquot in a 37 °C water-bath, and transfer to 1.5 ml screw cap tube from step 1. Mix well by pipetting a few times. Go directly to step A5 if the flow cytometry staining is not required.
Notes:- Keep samples on ice during the entire procedure since cell-associated RNA is very unstable.
- It is highly recommended to not thaw more than two samples at the time; the process before spinning at 8,000 x g cannot take more than 2 min in order to minimize cell-death and loss of cell-associated RNA.
- Keep samples on ice during the entire procedure since cell-associated RNA is very unstable.
- Flow staining:
- Mix the sample by pipetting up and down and transfer 50 µl (around 200,000 cells) to the prepared 96-well plate from step A2.
- Centrifuge the 96-well plate immediately at 500 x g at room temperature for 3 min.
- Remove supernatant carefully not disrupting cell pellet with multichannel pipette.
- Wash with 200 µl RPMI containing 20% FBS; then centrifuge at 500 x g at room temperature for 3 min.
- Remove supernatant carefully not disrupting cell pellets with multichannel pipette and resuspend in 25 µl staining buffer.
- Allow the cells to rest for 15 min room tempreture. In the meantime, proceed to the RNA/DNA co-extraction.
- Pre-mix 1 µl of CD3–APC, 1 µl CD4-FITC, 1 µl CD45–PerCP-Cy5.5 of antibodies per sample in a 1.5 ml screw-cap tube enough to run all the samples.
- Add 3 µl of the antibody-mix to each sample well.
- Incubate 15 min at room temperature in the dark.
- Add 175 µl staining-buffer to wash the cells.
- Centrifuge at 500 x g at room temperature for 3 min.
- Remove supernatant with multichannel and fix the cells with 200 µl of 1% FA. Mix by pipetting up and down and incubate at least 10 min to fix the cells.
- Acquire the stained samples in the Accuri flow cytometer. If the stained samples are not acquired immediately, store them at 4 °C until acquisition. Analysis should be performed by gating live PBMCs based on forward versus scatter profiles, followed by CD45+ for leukocyte identification. CD4 T cells are identified as double positive for CD3 andCD4 (CD3+ CD4+).
- Mix the sample by pipetting up and down and transfer 50 µl (around 200,000 cells) to the prepared 96-well plate from step A2.
- Centrifuge the 1.5 ml tubes at 8,000 x g at 4 °C for 2 min.
- Completely remove supernatant (with a sterile fine-tip transfer pipet) without disrupting the pellet.
- Add 350 µl RLT plus buffer + β-ME and mix thoroughly to homogenize the cells (vortex for 20 sec).
- Transfer the homogenized lysate to an All Prep DNA spin column (column with the purple ring) and centrifuge 1 min at >10,000 x g (max speed).
- Transfer the flow-through to the pink column for RNA purification (steps A11-17).
- Place the AllPrep DNA column in a clean 2 ml collection tube and store at 4 °C (or room temp for short periods) for DNA purification (steps A18-25).
Note: Do not freeze the All Prep DNA spin columns.
RNA purification- Extract RNA following the manufacturer’s protocol. Include a DNAse step to avoid any DNA-contamination.
- To increase RNA yield, heat the elution H2O (provided by the Allprep kit) at 60 °C in a heating block.
- For RNA elution, add 35 µl pre-heated H2O to the column and incubate 10 min at room temperature.
- Repeat elution using the eluate from step 13 at room temperature.
- Centrifuge 1 min at 10,000 x g.
- Measure RNA concentration by spectrophotometry (Nanodrop).
- Immediately proceed to cDNA generation discussed below or store RNA at -80 °C.
DNA purification- Extract DNA following the manufacturer’s protocol. To increase DNA yield, perform two sequential elutions (300 µl + 100 µl) with heated 10 mM Tris-HCl or EB Buffer (provided by the Allprep kit) at 60 °C.
- For the elution, add 300 µl of heated 10 mM Tris-HCl or EB buffer.
- Incubate for 10 min at room temperature.
- Centrifuge for 1 min at 10,000 x g at room temperature in an Eppendorf tube (provided by the Allprep Kit).
- Transfer the eluate into a 1.5 ml screw cap tube.
- Add 100 µl of 10 mM Tris-HCl or EB buffer (second elution).
- Centrifuge for 1 min at 10,000 x g in an Eppendorf tube.
- Transfer the eluate to the tube in step A21 for a final volume of 400 µl elution.
DNA precipitation- Add to the extracted DNA in this order:
- 1/10 volume of 3 M NaOAc (sodium acetate).
- 1 µl glycogen.
- 2 volumes of 100% ethanol.
- 1/10 volume of 3 M NaOAc (sodium acetate).
- Mix gently by inverting a few times. A DNA “cloud/ goop” should be visible. Store precipitated DNA samples overnight at -20 °C.
Note: If the DNA concentration is expected to be very low DNA can be stored at -80 °C for shorter incubations (at least 2 h) or overnight. - After cold incubation, centrifuge at >10,000 x g (max speed) for 30 min at 4 °C.
Note: Mark the orientation (north-side) of the tube to indicate where the DNA will pellet. - Discard the supernatant carefully with a sterile fine-tip transfer pipet without disturbing the DNA pellet.
- Wash DNA pellet with 1 ml 70% ethanol (freshly prepared). Mix gently.
- Centrifuge at >10,000 x g (max speed) for 30 min at 4 °C.
- Completely remove supernatant using a fine-tip transfer pipette. Do a quick spin. With a micro-pipette and sterile tip, remove any residual liquid without disrupting the pellet.
- Let tube air dry for 2 min or until dry.
Note: Do not over-dry the pellet. - Resuspend DNA pellet in 30 µl of 10 mM Tris-HCl or EB buffer (provided by kit from AllPrep Kit).
- Measure DNA concentration by spectrophotometry (Nanodrop).
- Stored at 4 °C (or -20 °C of long term storage).
- Quantification of HIV DNA [total and 2-LTR circles (long terminal repeat)] and human herpesvirus by ddPCR
We recommend running in multiplex CMV and EBV DNA, and total and 2-LTR circles HIV DNA (pol or gag). Since they are rare events, we recommend running them in triplicate. Also we use RPP30 (RNAse P) primer/probe set for host genomic DNA quantification (cell normalizer), which can be run in duplicate.
The protocol below describes reagents needed for a single PCR reaction: Scale up to the number of replicates x1.5 to account for sample loss due to pipetting. For example, if running triplicates, prepare DNA and reagents for 4.5 reactions.
Preparing DNA samples by restriction digestion- Each PCR reaction should contain a final concentration of DNA of ≤200 ng/µl in a total volume of 5 µl per well (maximum 1,000 ng/well). Dilute the samples with 10 mM Tris-HCl if necessary.
Notes:- If more sample needs to be tested, increase the number of reactions per well proportionally for restriction digestion and later divide into replicate wells for PCR.
- Do not use more than 1,000 ng of DNA per PCR reaction, as PCR reaction is greatly affected.
- If more sample needs to be tested, increase the number of reactions per well proportionally for restriction digestion and later divide into replicate wells for PCR.
- Prepare a digestion mix with the following components on ice. Quantities stated for 1 digestion reaction:
Digestion Concentration 1x 10x CutSmart buffer 1x 0.7 BanII restriction enzyme 10x U/µl 0.6 Molecular grade water 0.6 Subtotal 1.9 µl mix/well DNA sample 200x ng/µl 5 µl Total 6.9 Total vol/well - Add the appropriate volume of digestion mix (1.9 µl per PCR reaction) to each well containing DNA sample. Cover the wells containing sample with strip caps
- Incubate at 37 °C for 1 h in a thermocycler or a water bath.
- Centrifuge the 96-well plate at 200 x g for 1 min to collect any sample that might have condensed onto the cap or sides of the wells.
- Place the plate directly on ice while preparing the subsequent steps.
Quantitation of Total and 2-LTR circular HIV DNA, CMV and EBV DNA, and RPP30- To quantify RPP30, DNA samples need to be diluted 1:10 to get into the working range-100 ng of DNA is ideal. To do so:
- In a 96 well plate, aliquot 18 µl of nuclease-free molecular-grade water for each sample.
- Transfer 2 µl of digested DNA into the appropriate wells.
- Cap the plate and vortex briefly to mix.
- Centrifuge at 200 x g for 1 min and place on ice while preparing the subsequent steps.
- In a 96 well plate, aliquot 18 µl of nuclease-free molecular-grade water for each sample.
- Prepare a PCR mix with the following components on ice. Quantities stated for 1 PCR reaction:
1xRPP30 1x 2x ddPCR superMix for probes 10 20x RPP30 HEX primer/probe mix 1 Rnase/DNase free water 2.1 Total 13.1 µl Gag or Pol and 2-LTR multiplex 2x ddPCR superMix for probes 10 20x Gag or Pol HEX primer/probe mix 1 20x 2-LTR FAM primer/probe mix 1 RNase/DNase free water 1.1 Total 13.1 µl CMV and EBV multiplex 1x 2x ddPCR superMix for probes 10 20x EBV HEX primer/probe mix 1 20x CMV FAM primer/probe mix 1 RNase/DNase free water 1.1 Total 13.1 µl - Prepare a 96-well PCR clean plate on ice.
Note: When designing a PCR plate layout, it is better to group assay reactions in groups of 8 vertically, with any replicates spanning horizontally across the plate. This optimizes time and use of consumables. - For each PCR reaction, add 13.1 µl PCR mix to 6.9 µl non-diluted DNA sample target DNA and diluted 1:10 DNA for RRP30 quantification. Keep the replicates within the same well since they will be divided into separate wells for PCR at the droplet generation stage.
- For the subsequent droplet generation stage, 8 samples need to be loaded at any one time. If there are insufficient samples to ensure this, prepare the appropriate number of wells containing 20 µl of 1x buffer control solution.
- Seal the plate using an adhesive film and vortex lightly to ensure adequate mixing of sample and assay mix.
- Centrifuge at 200 x g for 1 min at room temperature to collect any sample that might have adhered to the top or sides of the wells.
- Proceed to droplet generation as described below.
Droplet generation
Note: For this section use low retention pipette tips to reduce pipette errors due to sample or assay mix sticking to plastic tips.- Turn on PCR plate sealer to allow it to come to proper temperature (180 °C).
- Pour 700 ml (1 container) of generator oil into a multichannel reagent trough.
- Load a cartridge into the droplet generator adaptor.
- Remove adhesive film from PCR plate. Using low retention tips, transfer 20 µl of PCR reaction to middle wells of cartridge. Use a P-20 multichannel for this process.
Note: 8 samples need to be loaded onto a cartridge at any one time. If there are any empty wells, the cartridge will not pressurize correctly and droplets will not form. - Transfer 70 µl of generator oil to the bottom wells of the cartridge. Use a P-200 multichannel for this process. Top wells are empty and will receive generated droplets.
- Hook generator gasket over generator adaptor and center, load into the droplet generator unit and run.
- When droplet generation is complete, remove adaptor from unit and discard rubber gasket. Top wells of cartridge will now contain droplets, with middle and lower wells nearly empty.
- Perform next step slowly and carefully as droplets are fragile and will shear if done too quickly. Avoid pressing and releasing plunger at maximum speed and smoothly draw or aspirate solution with pipette tips to ensure integrity of droplets. Set P-50 multichannel pipette to 40 µl and attach low retention tips; the plunger action on the P-50 is large enough to allow slow pipetting action to reduce shearing of the droplets. With the holder flat, position multichannel at ~80° angle so that pipette tips rest against the bottom of the cartridge. Slowly draw 40 µl of droplets from outlet wells.
- You should see a milky color in pipette tips; these are the formed droplets. Slowly pipette droplets into one column of 96-Well Semi-Skirted PCR plate. Do not go back to cartridge and re-pipette potentially remaining droplets; these droplets will become sheared and will affect downstream analysis.
- Repeat steps 18-24 as needed to fill the 96-well plate.
- Place a piece of sealing foil, glue side down, on top of the plate. Seal plate with pre-heated PCR plate sealer at 180 °C.
PCR- Use a thermocycler with the ability to set a 50% ramping speed (2-3 °C/sec) and has a heated lid. The chemistry of droplets requires a lower ramping speed to properly come to temperature for PCR reaction.
- Run the following thermocycler program:
gDNA - Gag or Pol and 2LTR multiplex and RPP30 95 °C 10 min 94 °C 30 sec 50 cycles 60 °C 60 sec 98 °C 10 min 4 °C ∞ gDNA – CMV and EBV 95 °C 10 min 94 °C 30 sec 50 cycles 54 °C 60 sec 98 °C 10 min 4 °C ∞ - When program is complete, store plate containing droplets at 4 °C for no more than 24 h or read immediately in Bio-Rad droplet reader.
Analysis
Template copies per sample are computed by averaging over all available replicate wells. Total cellular input is measured by halving the estimated number of RPP30 copies. Copy numbers of gag (or pol) or 2-LTR can be normalized to one million PBMCs as determined by RPP30 (total cell count) or to one million CD4 T cells (as determined by the percentage of CD4 T cells as measured by flow cytometry and multiplied by total cell count). CMV and EBV can be normalized to one million PBMCs as determined by RPP30 (total cell count). - Each PCR reaction should contain a final concentration of DNA of ≤200 ng/µl in a total volume of 5 µl per well (maximum 1,000 ng/well). Dilute the samples with 10 mM Tris-HCl if necessary.
- Quantification of cell associated HIV RNA (unspliced and multiply spliced) by ddPCR
We recommend running both unspliced (gag) and multiply spliced (Tat-Rev) RNA in a multiplex format. Poly-A, which quantifies fully elongated and correctly processed HIV-1 mRNA, should be run alone, not multiplexed. All RNA samples can be run in duplicate.
The protocol below describes reagents needed for a single PCR reaction: Scale up to the number of replicates x1.5 to account for sample loss due to pipetting. For example, if running duplicates, prepare 3 reactions.
Preparing RNA samples by reverse transcription- Determine the concentration of RNA (obtained from step A17 from section A) within samples to be tested by spectrophotometry (Nanodrop).
- RNA Prepare a 96-well PCR clean plate on ice. Into this, aliquot 15 µl RNA per PCR reaction.
- Prepare a reverse transcription (RT) mix using with the following components on ice from the iScript advanced cDNA synthesis Kit. Quantities are listed for 1 reaction.
RT-step 1x 20x iScript Advanced Reverse Transcriptase 1 5x iScript Advanced Reaction Mix 4 µl mix/well Subtotal 5 RNA 15 µl Total 20 total µl /well - Add the appropriate volume of RT mix (5 µl per PCR reaction) to each well containing RNA sample.
- Cover the wells containing sample with strip caps.
- Place the plate in a thermocycler and cycle with the following conditions:
RT-step 42 °C 30 min 85 °C 5 min 4 °C ∞ - Centrifuge the 96-well plate at 200 x g for 1 min at 4 °C to collect any sample that might have condensed onto the cap or sides of the wells.
- Place the plate place directly on ice.
- Dilute samples that contain >200 ng/µl cDNA (calculated as 2x the initial concentration of RNA, assuming 100% efficiency of RT reaction) using nuclease-free molecular-grade water.
Quantitation of HIV RNA (gag and Tat-rev)- Prepare a PCR mix with the following components (quantities stated for 1 reaction; to scale up, prepare mix for 1.1x the total number of reactions):
Gag- Tat/Rev multiplex 1x 2x ddPCR superMix for probes 10 20x Gag HEX 1 20x Tat/Rev FAM 1 RNase/DNase free water 0 Total 12 Poly-A 1x 2x ddPCR superMix for probes 10 20x Poly-A FAM 1 RNase/DNase free water 1 Total 12 - Prepare a 96-well PCR clean plate on ice.
Note: When designing a PCR plate layout, it is better to group assay reactions in groups of 8 vertically, with any replicates spanning horizontally across the plate. This optimizes time and use of consumables. - For each PCR reaction, add 12 µl PCR mix to 8 µl cDNA sample into each well of the PCR plate. Keep replicates within the same well, as they will be divided into separate wells for PCR at the droplet generation stage.
- For the subsequent droplet generation stage, 8 samples need to be loaded at any one time. If there are insufficient samples to ensure this, prepare the appropriate number of wells containing 20 µl of 1x buffer control solution.
- Seal the plate using an adhesive film and vortex lightly to ensure adequate mixing of sample and assay mix.
- Centrifuge at 200 x g for 1 min at room temperature to collect any sample that might have adhered to the top or sides of the wells.
- Proceed to droplet generation as described above (steps 15-26 from section B).
PCR- After droplet generation, use a thermocycler with the ability to set a 50% ramping speed (2-3 °C/sec) and has a heated lid. The chemistry of droplets requires a lower ramping speed to properly come to temperature for PCR reaction.
- Run the following thermocycler program:
cDNA 95 °C 10 min 94 °C 30 sec 60 cycles60 cycles 58 °C 60 sec 98 °C 10 min 4 °C ∞ - When program is complete, store plate containing droplets at 4 °C for no more than 24 h or read immediately in Bio-Rad Droplet Reader.
Analysis
Bio-Rad software calculates the concentration (copies/µl) for all samples.- For HIV DNA, we normalize data to million of PBMCs or CD4:
First, we calculate the number of PBMCs per well, using RPP30 results: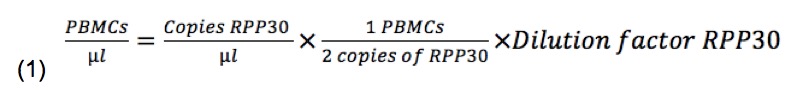
Then, for each transcript we calculate the copies per million PBMCs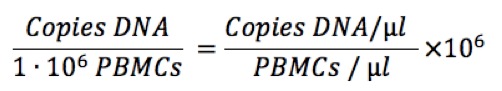
We can also normalize to one million CD4 T cells: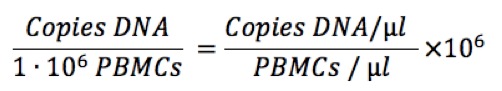
In equation (2), each percentage refers to the fraction positive for that marker within the prior cell population. Thus %CD45+ is the fraction within all PBMCs, %CD3+ is the fraction within all CD45+ cells, and %CD4 is measured among CD3+ cells. If cells are stained for these three markers, the percentages can be obtained by sequential gating on CD45→CD3→CD4.
Note that, for clinical samples, the %CD45+ is frequently unavailable. To allow accurate normalization to CD4 cells number, three-color flow cytometry should be performed directly. - For HIV RNA
Normalization for RNA is controversial. Since RNA expression may vary across cell types, disease stage, ART status, activation status, and other clinical variables, the use of a housekeeping gene is not routinely recommended for normalization of RNA in clinical samples. Here, we propose two alternative approaches for normalization of cellular HIV RNA, using RNA input, or using number of input cells (as measured by cell count or by RPP30 in the DNA fraction).
First, we need to calculate the total copies per well:
Data can be normalized to:- Total RNA
First, we need to calculate the amount of cDNA that we add to the reaction: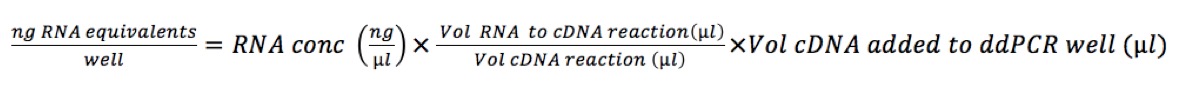
Then, we normalize to 100 ng of RNA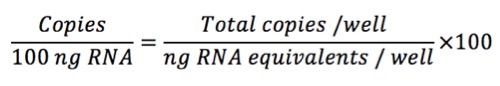
- Million of PBMCs
First, we need to calculate how many PBMCs are in the aliquot before extraction. We will use RPP30 from DNA assay to quantify the cell number: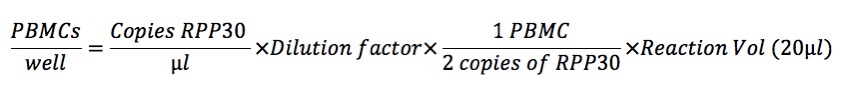
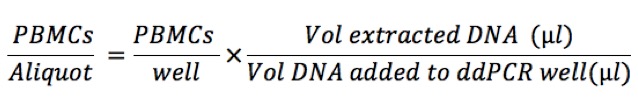
For each target RNA evaluated: Then, we normalize to millions of PBMCs:
Then, we normalize to millions of PBMCs:
We can also normalize to millions of CD4 T cells: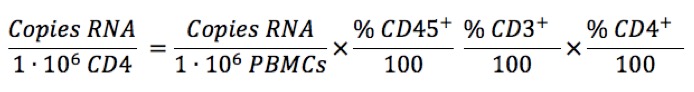
All of the calculations above use the output in copies/µl from the Bio-Rad reader software. If confidence intervals are desired, the same calculations can be duplicated for the values computed by the Bio-Rad software for the upper and lower confidence bound in copies / µl. This method typically provides a good estimate, because the cDNA copy number measurement is usually the greatest source of uncertainty among all the quantities used in these calculations.
Calculator template for HIV DNA and RNA (enclosed)
- Total RNA
- Determine the concentration of RNA (obtained from step A17 from section A) within samples to be tested by spectrophotometry (Nanodrop).
Representative data
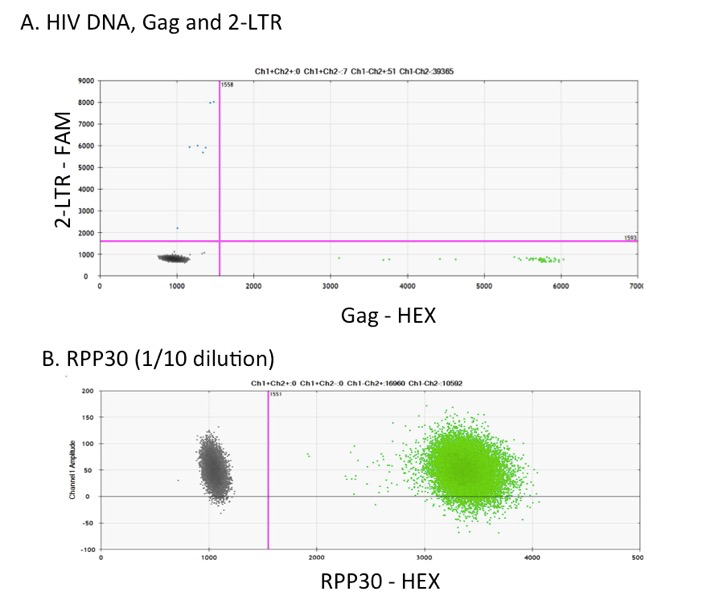
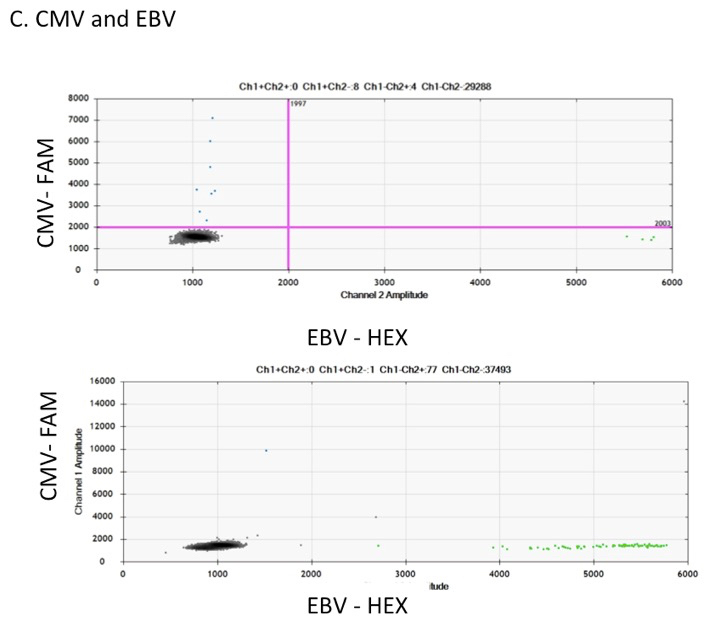
Figure 1. DNA assays for HIV, CMV and EBV. DNA extracted from PBMCs of HIV-infected ART-suppressed samples were ran in ddPCR. Examples of ddPCR dot plot analysis using Bio-Rad software of: A. HIV gag–HEX and 2-LTR–FAM in multiplex, B. RPP30–HEX diluted 1/10 and ran in singly, and (C) and CMV–FAM and EBV–HEX in multiplex.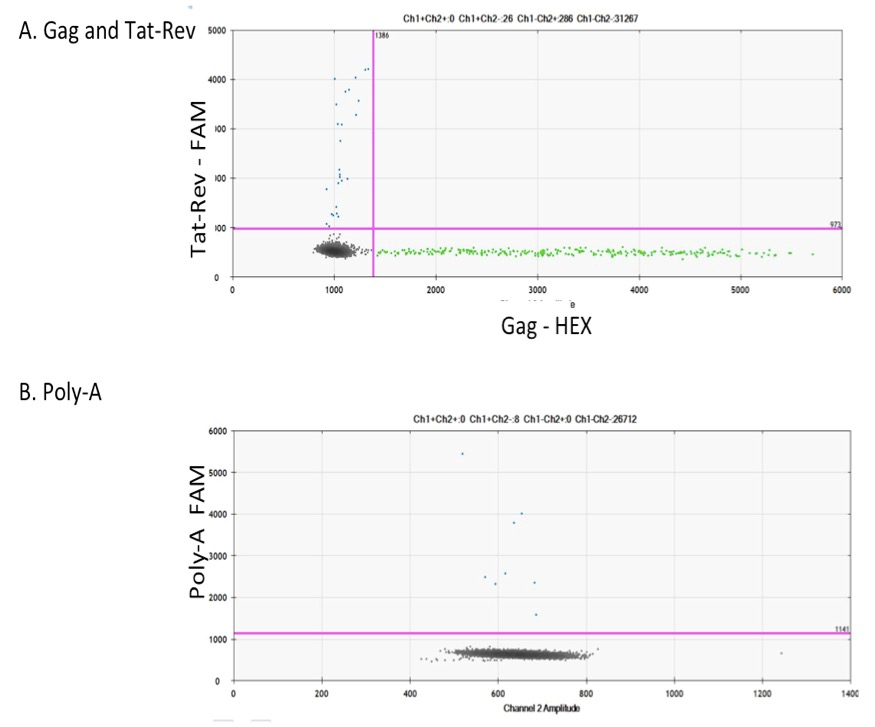
Figure 2. Cell-associated HIV RNA. RNA extracted from PBMCs of HIV-infected ART-suppressed samples were ran in ddPCR. Examples of ddPCR dot plot analysis using Bio-Rad software of :(A) HIV gag–HEX and Tat-Rec–FAM in multiplex and (B) Poly-A–FAM in singly.

Notes
- Restriction enzyme for HIV DNA: If other primers and probe set is used for HIV DNA, digest enzyme must be checked to avoid any cleavage in the new amplicon. Cleavage by endonucleases can be cross-referenced against the NEBcutter tool (http://tools.neb.com/NEBcutter).
Table 1. Primers and probe sets for ddPCRVirus Specimen Ref Reporter/ Quencher Dye Primer Forward Primer Reverse Probe HIV RPP30 HEX/ZEN/IBFQ GATTTGGACCTGCGAGCG GCGGCTGTCTCCACAAGT CTGACCTGAAGGCTCT Gag [2] HEX/ZEN/IBFQ AGTTGGAGGACATCAAGC
AGCCATGCAAATTGCTATGTCAGTTCCCCTT
GGTTCTCTAGACCATCAATGAGGAA
GCTGCAGAATGGGATPol [3] HEX/ZEN/IBFQ TACAGTGCAGGGGAAAGA
ATACTGCCCCTTCACCTTTCC TTTCGGGTTTATTACAGG
GACAGCAG2-LTR [4] FAM/ZEN/IBFQ AACTAGGGAACCCACTGC
TTAAGTCCACAGATCAAGGATAT
CTTGTCACACTACTTGAAGCACTC
AAGGCAAGCTTTmsTat-Rev [5] FAM/ZEN/IBFQ CTTAGGCATCTCCTATGG
CAGGAGGATCTGTCTCTGTCTCT
CTCTCCACCAGGGGACCCGACAGGCCC Poly-A [6] FAM/ZEN/IBFQ CAGATGCTGCATATAAGC
AGCTGTTTTTTTTTTTTTTTTTTTT
TTTTGAAGCACCTGTACTGGGTCTCTCTGG CMV CMV [7] FAM/ZEN/IBFQ AGGTCTTCAAGGAACTC
AGCAAGACGGCAATCGGTTTGTTGTAAAAACCCGTCAGCCATTCTCTCGGC EBV EBV [7] HEX/ZEN/IBFQ CGGAAGCCCTCTGGAC
TTCCCCTGTTTATCCGATGGAATG TGTACACGCACGAGAAATGCGCC
Recipes
- Staining flow cytometry buffer (staining buffer)
1x DPBS + 2% FBS sterile + 0.01% sodium azide - 1% formaldehyde (1% FA) buffer
1x DPBS + 1% formaldehyde - RLT plus buffer + β-ME
Prepare 1/100 BME in RLT plus buffer
For example, for 15 ml of RLT plus buffer add 150 µl of BME - 70% ethanol buffer
Mix 7 volumes of ethanol and 3 volumes of molecular grade water
Note: It is highly recommended to prepare fresh 70% ethanol for each extraction. - 10 mM Tris elution buffer
Dilute 1 M Tris to 10 mM Tris using molecular grade water - QIAGEN’s RNase-free DNase
Prepare according to manufacturer’s protocol prior to co-extraction
Use a syringe with detachable 20G needle. Pre-make aliquots enough to cover the number of reactions to-be-run and stored at -20 °C
Acknowledgments
This work was supported by the Department of Veterans Affairs, the UCSD Center for AIDS Research (P30 AI36214), the CARE Collaboratory (U19 AI096113) and the James B. Pendleton Charitable Trust. This protocol was adapted from previous work (Gianella et al., 2014, Strain et al., 2013).
References
- Gianella, S., Massanella, M., Richman, D. D., Little, S. J., Spina, C. A., Vargas, M. V., Lada, S. M., Daar, E. S., Dube, M. P. and Haubrich, R. H. (2014). Cytomegalovirus replication in semen is associated with higher levels of proviral HIV DNA and CD4+ T cell activation during antiretroviral treatment. J Virol 00831-00814.
- Christopherson, C., Kidane, Y., Conway, B., Krowka, J., Sheppard, H. and Kwok, S. (2000). PCR-Based assay to quantify human immunodeficiency virus type 1 DNA in peripheral blood mononuclear cells. J Clin Microbiol 38(2): 630-634.
- Rousseau, C. M., Nduati, R. W., Richardson, B. A., John-Stewart, G. C., Mbori-Ngacha, D. A., Kreiss, J. K. and Overbaugh, J. (2004). Association of levels of HIV-1-infected breast milk cells and risk of mother-to-child transmission. J Infect Dis 190(10): 1880-1888.
- Butler, S. L., Hansen, M. S. and Bushman, F. D. (2001). A quantitative assay for HIV DNA integration in vivo. Nat Med 7(5): 631-634.
- Schmid, A., Gianella, S., von Wyl, V., Metzner, K. J., Scherrer, A. U., Niederost, B., Althaus, C. F., Rieder, P., Grube, C., Joos, B., Weber, R., Fischer, M. and Gunthard, H. F. (2010). Profound depletion of HIV-1 transcription in patients initiating antiretroviral therapy during acute infection. PLoS One 5(10): e13310.
- Shan, L., Rabi, S. A., Laird, G. M., Eisele, E. E., Zhang, H., Margolick, J. B. and Siliciano, R. F. (2013). A novel PCR assay for quantification of HIV-1 RNA. J Virol 87(11): 6521-6525.
- Gianella, S., Morris, S. R., Anderson, C., Spina, C. A., Vargas, M. V., Young, J. A., Richman, D. D., Little, S. J. and Smith, D. M. (2013). Herpes viruses and HIV-1 drug resistance mutations influence the virologic and immunologic milieu of the male genital tract. AIDS 27(1): 39-47.
- Vargas-Meneses, M. V., Massanella, M., Ignacio, C. C. and Gianella, S. (2015). Quantification of HIV RNA and human herpesvirus DNA in seminal plasma. Bio-protocol 5(9): e1465.
- Strain, M. C., Lada, S. M., Luong, T., Rought, S. E., Gianella, S., Terry, V. H., Spina, C. A., Woelk, C. H. and Richman, D. D. (2013). Highly precise measurement of HIV DNA by droplet digital PCR. PLoS One 8(4): e55943.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Massanella, M., Gianella, S., Lada, S. M., Richman, D. D. and Strain, M. C. (2015). Quantification of Total and 2-LTR (Long terminal repeat) HIV DNA, HIV RNA and Herpesvirus DNA in PBMCs. Bio-protocol 5(11): e1492. DOI: 10.21769/BioProtoc.1492.
Category
Microbiology > Microbial genetics > RNA > dd-PCR
Microbiology > Microbial genetics > DNA > DNA quantification
Microbiology > Microbial genetics > DNA > DNA detection and isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.