- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation of Multiplexed Small RNA Libraries from Plants

Published: Vol 4, Iss 21, Nov 5, 2014 DOI: 10.21769/BioProtoc.1275 Views: 14426
Original research article
The authors used this protocol in:

May 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols



Abstract
High-throughput sequencing is a powerful tool for exploring small RNA populations in plants. The ever-increasing output from an Illumina Sequencing System allows for multiplexing multiple samples while still obtaining sufficient data for small RNA discovery and characterization. Here we describe a protocol for generating multiplexed small RNA libraries for sequencing up to 12 samples in one lane of an Illumina HiSeq System single-end, 50 base pair run. RNA ligases are used to add the 3’ and 5’ adaptors to purified small RNAs; ligation products that lack a small RNA molecule (adaptor-adaptor products) are intentionally depleted. After cDNA synthesis, a linear PCR step amplifies the DNA fragments. The 3’ PCR primers used here include unique 6-nucleotide sequences to allow for multiplexing up to 12 samples.
Keywords: Small RNAMaterials and Reagents
- Low molecular weight gel-based small RNA isolation
- ssRNA marker (Takara Bio Company, catalog number: 3416 )
- Reagents for 17% polyacrylamide, 7M Urea PAGE
- 37.5:1 polyacrylamide:bisacrylamide (see Recipes)
- Urea (Life Technologies, catalog number: 15505-050 )
- TEMED (Life Technologies, catalog number: 15524-010 )
- 10% ammonium persulfate (Sigma-Aldrich, catalog number: A3678-100G ) (see Recipes)
- 37.5:1 polyacrylamide:bisacrylamide (see Recipes)
- 2x RNA loading dye (see Recipes)
- ssRNA marker (Takara Bio Company, catalog number: 3416 )
- DE81-based nucleic acid purification
- Fiber glass (Corning Incorporated, catalog number: 988-10144 )
- Extra thick blot paper (Bio-Rad Laboratories, catalog number: 170-3969 )
- Whatman DE81 ion exchange cellulose chromatography paper (Thermo Fisher Scientific, catalog number: 05-717-1A )
- GlycoBlue coprecipitant (Life Technologies, catalog number: AM9516 )
- 100% ethanol
- 75% ethanol
- EB buffer (QIAGEN, catalog number: 19086 )
- Qubit RNA HS Assay Kit for quantification of purified small RNAs (Life Technologies, catalog number: Q32852 )
- Qubit dsDNA HS Assay Kit for quantification of DNA amplicon (Life Technologies, catalog number: Q32854 )
- Low salt buffer (see Recipes)
- High salt buffer (see Recipes)
- Fiber glass (Corning Incorporated, catalog number: 988-10144 )
- Multiplexed small RNA libraries preparation
- 8-Strip PCR thin-walled, 200 µl tubes (Corning Incorporated, Axygen®, catalog number: PCR-0208-CP-C )
- miRNA cloning linker 1 (/5'App/CTGTAGGCACCATCAAT/3'ddC/; 1 nm) (Integrated DNA Technologies, catalog number: 11-04-03-05 )
- Truncated, K227Q mutation T4 RNA ligase 2 (New England Biolabs, catalog number: M0351L )
- De-adenylase (New England Biolabs, catalog number: M0331S )
- Exonuclease VII (United State Biological, catalog number: 70082Z )
- RNA 5’ adapter (GUUCAGAGUUCUACAGUCCGACGAUC) (Illumina RA5, catalog number: 15013205 )
- dATP (Life Technologies, catalog number: 55082 )
- T4 RNA ligase I (Life Technologies, catalog number: AM2141 )
- RT-PCR primer (ATTGATGGTGCCTACAG; 25 nmol; de-salted) (Integrated DNA Technologies)
- SuperScript III (Life Technologies, catalog number: 18080051 )
- Phusion high fidelity II (Thermo Fisher Scientific, catalog number: F549L )
- 5' PCR primer (Illumina small RNA PCR primer 2: AATGATACGGCGACCACCGACAGGTTCAGA-GTTCTACAGTCCGA)
- 3' indexed PCR primer I1 - I12 (100 nmol; PAGE-purified) (Integrated DNA Technologies)
Note: The barcodes below (underlined sequences) are reverse complemented in the final Illumina output sequence (see sequence in square brackets for each primer).
I1 [CGATGT]: CAAGCAGAAGACGGCATACGAACATCGATTGATGGTGCCTACAG
I2 [GATCAC]: CAAGCAGAAGACGGCATACGAGTGATCATTGATGGTGCCTACAG
I3 [CAGATG]: CAAGCAGAAGACGGCATACGACATCTGATTGATGGTGCCTACAG
I4 [TACGTT]: CAAGCAGAAGACGGCATACGAAACGTAATTGATGGTGCCTACAG
I5 [TTACCA]: CAAGCAGAAGACGGCATACGATGGTAAATTGATGGTGCCTACAG
I6 [ACTGTA]: CAAGCAGAAGACGGCATACGATACAGTATTGATGGTGCCTACAG
I7 [ATCACG]: CAAGCAGAAGACGGCATACGACGTGATATTGATGGTGCCTACAG
I8 [ACTTGT]: CAAGCAGAAGACGGCATACGAACAAGTATTGATGGTGCCTACAG
I9 [GCCAAT]: CAAGCAGAAGACGGCATACGAATTGGCATTGATGGTGCCTACAG
I10 [TGCTAG]: CAAGCAGAAGACGGCATACGACTAGCAATTGATGGTGCCTACAG
I11 [CTTGTA]: CAAGCAGAAGACGGCATACGATACAAGATTGATGGTGCCTACAG
I12 [TCAGGC]: CAAGCAGAAGACGGCATACGAGCCTGAATTGATGGTGCCTACAG
- QIAquick PCR Purification Kit (QIAGEN, catalog number: 28106)
- Diethylpyrocarbonate (DEPC)-H2O/RNase-free H2O
- DNA size marker suitable for detecting a ~120 bp fragment (50 bp step ladder, Promega Corporation, catalog number: G4521 ; 25 bp step ladder, Promega Corporation, catalog number: G4511 )
- 3 M sodium acetate (NaOAc) (pH 5.5)
- Reagents for 6% native PAGE
- 37.5:1 polyacrylamide:bisacrylamide (see Recipes)
- TEMED (Life Technologies, catalog number: 15524-010)
- 10% ammonium persulfate (Sigma-Aldrich, catalog number: A3678-100G) (see Recipes)
- 37.5:1 polyacrylamide:bisacrylamide (see Recipes)
- 8-Strip PCR thin-walled, 200 µl tubes (Corning Incorporated, Axygen®, catalog number: PCR-0208-CP-C )
Equipment
- Thermocycler
- Microcentrifuge: For use at room temperature and at 4 °C
- Fisher gel-loading tips (Thermo Fisher Scientific, catalog number: 02-707-182 )
- Agarose gel electrophoresis systems (Bio-Rad Laboratories, Sub-Cell GT system)
- Polyacrylamide gel electrophoresis system (Thermo Fisher Scientific, owl Dual-Gel vertical electrophoresis systems)
- 65 °C water bath
- Semi-dry blotting system (Sigma-Aldrich, semi-dry blotter, catalog number: Z340502 )
- Heating block
Procedure
Part I. Low molecular weight gel-based small RNA isolation
Notes:
- This step is not required if you already purified your small RNAs (e.g. by immunoprecipitation). See note A in Notes section. In this case, skip to section C.
- Duration of this step:
- Gel preparation and polymerization: 1+ h.
- Gel pre-run time: 30+ min.
- Gel running time: 4+ h, plus loading time and staining.
- Gel preparation and polymerization: 1+ h.
- Preparing a 17% polyacrylamide gel containing 7 M Urea in 0.5x TBE
- The day before you plan to run the gel, assemble glass plates and cover with Saran wrap to protect.
- Use plates of 14 cm x 16 cm and 1.5 mm spacers.
- Clean all materials (plates, spacers and comb).
- Wash with 10% sodium dodecyl sulfate (SDS).
- Rinse thoroughly with distilled-water.
- Rinse with 95% ethanol.
- Allow to air-dry.
- Wash with 10% sodium dodecyl sulfate (SDS).
- Use plates of 14 cm x 16 cm and 1.5 mm spacers.
- While preparing polyacrylamide gel mix (below): Seal the outside edges of your plate set-up with 2% agarose (55 °C) to prevent leaking. Sealing is achieved in a few minutes.
- Prepare the mix (30 ml) for making one 17% denaturing Urea-PAGE/0.5x TBE gel with 15-well comb by combining the following:
Reagent (stock concentration)
Volume (final concentration)
Acrylamide:bis 37.5:1 solution (30%)
17 ml (17%)
Urea
12.6 g (7 M)
TBE (10x)
1.5 ml (1x)
H2O
2.0 ml
- Mix the above together thoroughly by inversion.
- Heat to 65 °C for 10 min to dissolve the urea; invert to mix every ~2 min to help the process.
- Let stand on bench for ~10 min then transfer to an ice bath in the cold room (4 °C) for 15+ min.
- Add the remaining reagents:
Reagent (stock concentration)
Volume (final concentration)
TEMED
25 µl *Mix thoroughly by inversion
APS (10%)
150 µl (0.05%) *Mix thoroughly by inversion
- Pour the gel and allow it to polymerize for at least 30 min; should see beginning of polymerization within ~4 min.
- Assemble your electrophoresis device and add the running buffer (0.5x TBE) to inside and outside chambers.
- Rinse out wells with a syringe; rinse away bubbles along bottom edge of glass plates.
- Gel must be pre-run (at 180 V) for 30-60 min to remove all traces of APS (ammonium and persulfate ions), to distribute/equilibrate any stabilizing factors or ions that were added to the running buffer, and to ensure a constant gel temperature during the run of the samples. Occurrence of concave front of bands is reduced when pre-running the gel. If desired, a blank solution can be loaded: Mix equal amounts of DEPC-H2O and 2x loading dye. 15 µl/lane is sufficient.
- The day before you plan to run the gel, assemble glass plates and cover with Saran wrap to protect.
- Preparing RNA samples and running the gel
*Keep sample volume to a minimum for best resolution (≤50 µl).- Prepare samples, markers, ladders and blanks in equal volume using 2x RNA loading dye at a final concentration of 1x.
- Heat samples at 65 °C for 10 min and immediately quench on ice. Spin briefly to collect sample at the bottom of tube.
- Load samples.
Note: Before loading samples clean the lanes with a syringe to remove the excess of urea. Sample loading determines the ultimate quality of your gel. We use Fisher gel-loading tips to load small RNA gels. Load the sample at the very bottom of the well in a flat band, no triangles, blobs, or other sloppy loading.
- Run at 180 V in 0.5x TBE buffer until the bromophenol blue (BPB) dye reaches the bottom of the gel (~4 to 4.5 h).
- Stain gel with ethidium bromide in 0.5x TBE buffer.
- See below for eluting RNA from acrylamide fragment using DE81 paper.
- Prepare samples, markers, ladders and blanks in equal volume using 2x RNA loading dye at a final concentration of 1x.
Part II. DE81-based nucleic acid purification
Notes: Duration of this step
- Excision of gel slices and semi-dry transfer: 40+ min.
- Elution (RNA): 60+ min (day 1); precipitation #1: overnight; precipitation #2: 2.5+ h.
- Elution (DNA): 60+ min (day 1); precipitation #1: 1+ h; precipitation #2: 2+ h.
- Prepare elution tubes
- Remove the cap from a 500 µl microcentrifuge tube and poke a hole in the bottom center of the tube with a 25-gauge needle (Figure 1A). Label the tube.
- Stuff ~25-40 mg of sterile glass wool in the bottom (not through the hole) of the tube using a sterile toothpick or pipette tip. Compact the glass wool to just below the 0.1 ml graduation mark of the tube to leave room for the DE81 paper and buffer (Figure 1B).
- Remove the cap from a 2 ml microcentrifuge tube (Figure 1C) and place the prepared 500 µl tube (Figure 1D) inside.
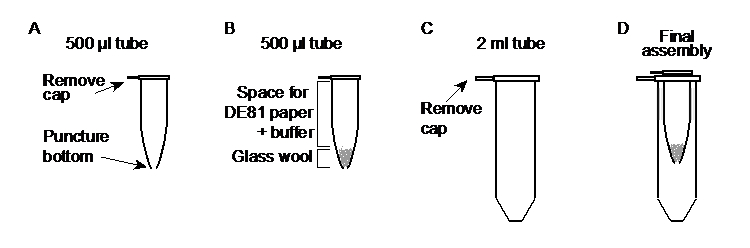
Figure 1. Preparation of the elution tubes. A. A 500 µl microcentrifuge tube has the cap removed and a hole made in the bottom center. B. Glass wool is placed inside the tube and compacted, leaving space for the buffer and DE81 paper. C. A 2 ml microcentrifuge tube has the cap removed. D. The prepared 500 µl tube is placed inside the 2 ml tube and is ready for use.
- Remove the cap from a 500 µl microcentrifuge tube and poke a hole in the bottom center of the tube with a 25-gauge needle (Figure 1A). Label the tube.
- Nucleic acid excision and transfer
- Place one thick Whatman paper, pre-soaked with 0.5x TBE, on semi-dry blotter; roll with a test tube or 10 ml pipet to remove any air bubbles.
- Excise desired fragment from gel (Figure 2) and place on a piece of DE81 paper labeled using a pencil. Each sample/replicate should be placed onto its own DE81 paper. DE81 paper contains diethylaminoethyl (DEAE) cellulose functional groups that will bind nucleic acids, which can be reversed by the addition of a high salt buffer (Dretzen et al., 1981).
Include a transfer control - i.e. transfer a gel fragment containing bands from the Ladder lane.
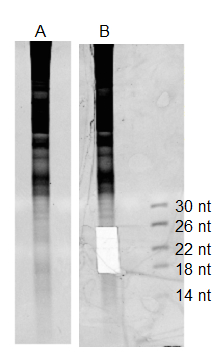
Figure 2. 17% Polyacrylamide-Urea gel for small RNA isolation stained with ethidium bromide. A. Total RNA before excision of small RNAs. B. Same lane, with small RNAs (18 - 26 nt) excised. ssRNA marker sizes are indicated.
- Place paper + gel onto thick Whatman paper on blotter.
- Place a second piece of pre-soaked thick Whatman paper on top.
- Carefully roll the top of the second Whatman paper to remove any air bubbles.
- Transfer 20 min at 400 mAmp in semi-dry blotter.
- Remove the top Whatman paper.
- Verify transfer by examining the gel fragment and DE81 paper of the marker of the transfer control, side by side, on a UV trans-illuminator. Nucleic acid should not be visible in the gel fragment (Figure 3A) but should be visible on the DE81 paper (Figure 3B). Transfer of ssRNA markers can be difficult to visualize.
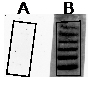
Figure 3. Verification of the nucleic acid transfer. A. Gel fragment is outlined and no nucleic acid is visible. B. DE81 paper with visible nucleic acid transferred (50 bp DNA step ladder).
- While the gel + paper is still on the bottom Whatman paper, use a razor to trim away the excess DE81 paper. Discard gel slice and carefully stuff the DE81 paper into the elution tube containing glass wool.
- Verify transfer by examining the gel fragment and DE81 paper of the marker of the transfer control, side by side, on a UV trans-illuminator. Nucleic acid should not be visible in the gel fragment (Figure 3A) but should be visible on the DE81 paper (Figure 3B). Transfer of ssRNA markers can be difficult to visualize.
- Place one thick Whatman paper, pre-soaked with 0.5x TBE, on semi-dry blotter; roll with a test tube or 10 ml pipet to remove any air bubbles.
- Nucleic acid elution
- Thoroughly submerge DE81 paper with ~200 µl (usually 200-500 µl) of low salt buffer. Use pipet tip to gently push paper as low as possible so it is completely covered by the buffer.
- Spin 30 sec at maximum speed in a tabletop microcentrifuge. Discard flow-through.
- Repeat low salt buffer wash twice for a total of three washes.
- Transfer upper column/paper assembly to a clean, labeled 2 ml tube with cap removed.
- Elution:
- Add 150 µl high salt buffer to paper, and submerge it.
- Incubate at 70 °C for 15 min - use foil to cover the tubes. Do not use a water bath.
- Spin briefly (10 sec) in a tabletop microcentrifuge and save flow through.
- Add 150 µl high salt buffer to paper, and submerge it.
- Place a new 2 ml tube beneath filter assembly and repeat step C5 (elution) twice more.
- Ethanol precipitation:
- Combine the three eluates and add 20 µg glycogen (e.g. GlycoBlue).
- Add 2.5 volumes 100% ethanol and mix. Ideally the precipitation volumes fit in a single 1.5 ml microcentrifuge tube. Using 150 µl of high salt buffer + ~1,150 µl ethanol will just fit in a 1.5 ml microcentrifuge tube = more efficient precipitation.
- Precipitate at -80 °C: Overnight for RNA samples; 1+ h for DNA samples.
- Centrifuge at maximum speed in a refrigerated microcentrifuge:
- 35 min for RNA samples.
- 15 min for DNA samples.
- 35 min for RNA samples.
- Pour off ethanol and wash pellet with 75% ethanol.
- Spin 5 min at maximum speed in refrigerated microcentrifuge.
- Remove ethanol and drain tube upside-down on a clean KimWipe for 10 min. If necessary, use a clean KimWipe to remove excess liquid from the inside of the tube - avoid touching the (blue) pellet.
- Resuspend pellet in 40 µl DEPC-treated water (if multiple pellets for one sample, pool pellets at this step into a final volume of 40 µl).
- Combine the three eluates and add 20 µg glycogen (e.g. GlycoBlue).
- A second precipitation is required to remove residual salt for downstream enzymatic steps:
- Add 4 µl 3 M NaOAc. Add 100 µl 100% ethanol. Mix. Precipitate at -80 °C for at least 2 h (or overnight)
- Follow steps C7d-g.
- Resuspend final pellet in 10-12 µl of DEPC-H2O for RNA or 10-12 µl of EB buffer for DNA.
- Follow steps C7d-g.
- Add 4 µl 3 M NaOAc. Add 100 µl 100% ethanol. Mix. Precipitate at -80 °C for at least 2 h (or overnight)
- If isolating small RNAs: RNA is now ready to be a template for the Multiplexed small RNA libraries preparation step (section C below).
- Quantification: 2 µl + Qubit RNA HS Assay Kit. For samples where the Qubit returns a value for the small RNAs, use the recommended amount (5-10 µg) for small RNA library preparation. For samples that are too low to detect, we recommend using the entire sample (up to 11.5 µl) as the input for library preparation.
- If isolating DNA: DNA is now ready for quantification and submission to sequencing facility.
- Quantification: 2 µl + Qubit dsRNA HS Assay Kit.
- Quantification: 2 µl + Qubit dsRNA HS Assay Kit.
- Thoroughly submerge DE81 paper with ~200 µl (usually 200-500 µl) of low salt buffer. Use pipet tip to gently push paper as low as possible so it is completely covered by the buffer.
Part III. Multiplexed small RNA libraries preparation
Notes:
- For small RNAs from Total RNA: Recommend 5-10 ng of small RNAs (i.e. isolated from 50 µg of Total RNA).
- Protocol has been successfully used to multiplex 2, 3, 4, 5, 6, 10 or 12 samples in one flow-cell lane.
- Typical levels of adaptor-adaptor contamination in the Illumina data are <1%.
- Duration of this step:
- Step 1 is an overnight step.
- Steps 2-7 can be performed in 1 day, with step 8 plus DE81 elution on the second day.
- Step 1 is an overnight step.
- Left over RNA ligation products and cDNA products can be saved at -20 ºC to be used to create additional DNA amplicons if the final concentration is insufficient for sequencing.
- Mix and spin refers to a brief vortex followed by a brief centrifugation.
- 3' adaptor ligation (ATP-independent)
- Set up the ligation reaction in a sterile, nuclease free 200 µl PCR tube on ice for each sample using the following:
Reagent (stock concentration)
Volume (final concentration)
3’ Adaptor (10 µM)
1.5 µl (1.3 µM)
RNA (variable)
Variable (10 ng)
H2O
Final volume: 11.5 µl
- Mix and spin samples.
- Incubate the tubes in a pre-heated thermal cycler at 70 °C for 2 min and then place the tubes on ice.
Note: It is very important to keep the mixture on ice after the 70 °C incubation to prevent secondary structure formation.
- Pre-cool the thermocycler to 16 °C.
- Prepare the following master mix on ice (1x):
Reagent (stock concentration)
Volume (final concentration)
Ligation buffer (10x)
1.5 µl (1x)
RNaseOUT (40 U/µl)
1 µl (40 U)
T4 RNA ligase 2, truncated, K227Q (200 U/µl)
1 µl (200 U)
- Add 3.5 µl of the master mix to each tube. Mix and spin.
- Incubate the tubes in the pre-cooled thermocycler at 16 °C overnight (15-18 h). See note B in Notes section.
- Set up the ligation reaction in a sterile, nuclease free 200 µl PCR tube on ice for each sample using the following:
- 3' adaptor de-adenylation
- To each Ligation reaction, add 1 µl of De-adenylase (20 U/µl). Mix and spin.
- Incubate the tube in the thermocycler at 30 °C for 15 min.
- To each Ligation reaction, add 1 µl of De-adenylase (20 U/µl). Mix and spin.
- 3' adaptor digestion
- To each Ligation reaction, add 1 µl of Exonuclease VII (10 U/µl). Mix and spin.
- Incubate the tube in the thermocycler at 37 °C for 15 min.
- To each Ligation reaction, add 1 µl of Exonuclease VII (10 U/µl). Mix and spin.
- 5' adaptor ligation (ATP-dependent)
- Aliquot the required amount of the 5’ RNA adaptor (1 µl/reaction) into a thin-walled 200 µl PCR tube then incubate at 70 °C for 2 min and chill on ice.
- In this tube, prepare a Master Mix on ice using the following (1x):
Reagent (stock concentration)
Volume (final concentration)
5’ RNA adaptor (15 pmol/µl)
1 µl (15 pmol)
dATP (10 mM)
1 µl (0.48 mM)
T4 RNA ligase (5 U/µl)
1 µl (5 U)
H2O
1 µl
- Add 4 µl of the master mix to each ligation tube. Mix and spin.
- Incubate at 28 °C for 1 h, and then place the tube on ice.
- Aliquot the required amount of the 5’ RNA adaptor (1 µl/reaction) into a thin-walled 200 µl PCR tube then incubate at 70 °C for 2 min and chill on ice.
- cDNA synthesis
- Label a new tube for each sample. Transfer 6 µl of the ligation product to the new tube.
- Prepare a master mix on ice using the following (1x):
Reagent (stock concentration)
Volume (final concentration)
RT-PCR primer (100 µM)
2.5 µl (5 µM)
dNTPs (10 mM)
2.5 µl (0.5 mM)
H2O
18 µl
- Add 23 µl of the master mix to each tube. Mix and spin.
- Heat tubes to 65 °C for 5 min in thermocycler. Cool on ice for at least 2 min.
- Prepare a second master mix with the following (1x):
Reagent (stock concentration)
Volume (final concentration)
RT buffer (10x)
5 µl (1x)
MgCl2 (25 mM)
10 µl (5 mM)
DTT (100 mM)
5 µl (10 mM)
RNaseOUT (40 U/µl)
1 µl (40 U)
- Add 21 µl of the master mix to each sample (final volume in each tube: 50 µl). Mix and spin.
- Incubate tubes on bench top for 2 min to allow for primer-RNA hybridization.
- Add 1 µl SuperScript III RT polymerase (200 U/µl) to each tube. Mix and spin. Transfer to thermocycler:
a.Polymerization reaction: 50 min at 50 °C.
b.Inactivate reaction: 5 min at 85 °C.
- Cool the tubes to 4 °C, spin them briefly, and then add 1 µl of RNase H (2 U). Mix well and spin. Transfer to thermocycler:
a.37 °C for 20 min.
- Cool samples on ice.
- Label a new tube for each sample. Transfer 6 µl of the ligation product to the new tube.
- Linear PCR Amplification
- Label a new set of tubes. Transfer 25 µl of the cDNA reaction product to the new tubes.
- Prepare a master mix on ice using the following (1x):
Reagent (stock concentration)
Volume (final concentration)
Phusion HF buffer (5x)
10 µl (1x)
5’ PCR primer (25 µM)
0.5 µl (0.25 µM)
3’ indexed PCR primer (25 µM)
0.5 µl (0.25 µM)
dNTP Mix (10 mM)
0.5 µl (0.1 mM)
Phusion polymerase (2 U/µl)
0.5 µl (1 U)
H2O
13 µl
- Aliquot 25 µl of the master mix to each tube. Mix and spin.
- Recommended PCR conditions:
- 30 sec at 98 °C
- Up to 17 cycles of: 10 sec at 98 °C; 30 sec at 60 °C; 15 sec at 72 °C
- 10 min at 72 °C
- Hold at 4 °C
- 30 sec at 98 °C
- Label a new set of tubes. Transfer 25 µl of the cDNA reaction product to the new tubes.
- Verification Gel
- Pour a 2% agarose gel with 20-well comb. Mix 5 µl of PCR product + 5 µl of loading dye (preferably Orange G or a dye that will not obscure a band at ~100 bp). Load all 10 µl on gel and run at 80-85 V for ~20 min. Include a DNA ladder that has a 100 bp band (e.g. 50 bp step ladder).
- While agarose gel is running, clean up the PCR reactions using Qiagen QIAquick PCR Purification Kit.
- Add 5 µl of 3 M NaOAc (pH 5.5) to PCR sample + buffer PB to adjust the pH.
- Elute with 30 µl of EB.
- Add 5 µl of 3 M NaOAc (pH 5.5) to PCR sample + buffer PB to adjust the pH.
- Visualize agarose gel - the desired band should be just above 100 bp (Figure 4). Expected size of successful amplicons = 112-120 bp (5’Adaptor: 49 bp, 3’Adaptor: 45 bp, small RNA: 18-26 bp) (Figure 4A). Adaptor-adaptor products are 94 bp (Figure 4B).

Figure 4. Agarose check gel with 50 bp step ladder. A. Amplicon band is the desired size, above 100 base pairs. B. Sample band is smaller than expected due to the excess of adaptor-adaptor product.
- Pour a 2% agarose gel with 20-well comb. Mix 5 µl of PCR product + 5 µl of loading dye (preferably Orange G or a dye that will not obscure a band at ~100 bp). Load all 10 µl on gel and run at 80-85 V for ~20 min. Include a DNA ladder that has a 100 bp band (e.g. 50 bp step ladder).
- Purification gel
- Prepare 6% native PAGE/0.5x TBE gel with 15-well comb by combining the following:
Reagent (stock concentration)
Volume (final concentration)
Acrylamide: bis 37.5:1 solution (30%)
6 ml (6%)
TBE (10x)
1.5 ml (0.5x)
H2O
22.2 ml - Mix the above together thoroughly by inversion.
- Add the remaining reagents:
Reagent (stock concentration)
Volume (final concentration)
TEMED
33 µl
APS (10%)
240 µl (0.08%)*Mix thoroughly by inversion
- Pour gel into assembled glass plates and allow to polymerize for at least 30 min.
- Mix the above together thoroughly by inversion.
- Prepare samples: Add BPB dye in a 2 (sample):1 (BPB) ratio (i.e. 30 µl of sample to 15 µl loading buffer).
- Load 15 µl/lane (for a total of 3 lanes / sample).
- This is to prevent overloading one lane, which leads to smearing.
- Load 15 µl/lane (for a total of 3 lanes / sample).
- Use 1 µl/lane of 25 bp step ladder or 1 µl/lane of 50 bp step ladder for markers in a final volume of 15 µl.
- Run gel at 100 V until BPB runs off the bottom edge of the gel (~2.5 to 3 h).
- Stain with SybrGold (35 µl in 100 ml 0.5x TBE) for at least 10 min. Carefully excise the bands corresponding to DNA amplicons (Figure 5), avoiding any adaptor-adaptor bands. Use a different razor for each sample. Clean up DNA using DE81 transfer method (see section B). When multiple lanes are used for one sample, transfer all lanes onto the same piece of DE81 paper.
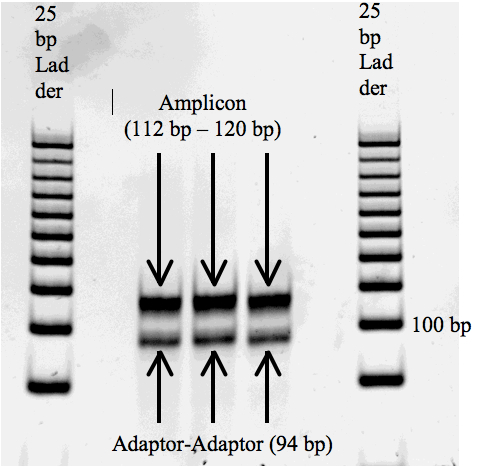
Figure 5. DNA amplicon purification gel. Desired amplicons are indicated (above 100 bp marker). Undesired adaptor-adaptor products are also present but at a reduced level compared to the DNA amplicons.
- Prepare 6% native PAGE/0.5x TBE gel with 15-well comb by combining the following:
- Sample multiplexing
- In order to submit small RNA libraries to a sequencing center as a multiplexed sample, the individual samples must be mixed together. After quantification of each library preparation using the Qubit dsDNA HS Assay Kit, calculate the approximate nanomolar (nM) concentration of each sample. The following formula can be used:
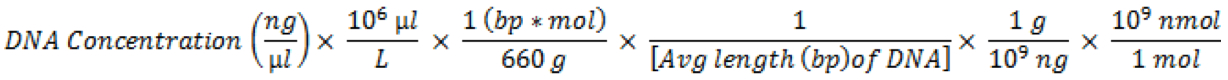
Libraries can now be mixed together in equal nanomolar amounts. The final concentration of the mixed sample will depend on the specific requirements of the sequencing center.
- In order to submit small RNA libraries to a sequencing center as a multiplexed sample, the individual samples must be mixed together. After quantification of each library preparation using the Qubit dsDNA HS Assay Kit, calculate the approximate nanomolar (nM) concentration of each sample. The following formula can be used:
Notes
- In Carbonell et al. (2012), small RNA libraries were constructed from Total RNA and small RNA that co-immunoprecipitated with Arabidopsis thaliana ARGONAUTE1. For co-immunoprecipitated small RNA, the isolation of small RNA by gel electrophoresis (procedure A) was skipped, and samples were used directly in procedure C.
- This protocol has been used to prepare multiplexed small RNA libraries from fungi (unpublished results) and should be valid for other organisms. We recommend reducing the 3’ adaptor ligation incubation time to 1-2 h for samples from organisms with small RNAs that lack 2’-O-methylation at their 3’ end (Munafó and Robb, 2010).
- After sequencing, data should be available in FASTQ format. In order to use the small RNA data in downstream applications, the raw FASTQ data need to be de-multiplexed (identification of the sample index sequence) and 3’ adaptor trimmed (identification and removal of miRNA cloning linker 1). We provide the script LibParse.pl (https://github.com/carringtonlab/srtools) to processes small RNA data sets generated using the protocol described here. LibParse.pl outputs small RNA sequences in FASTA format with read counts for each unique sequence. Additional scripts in the SRTOOLS repository are designed to utilize LibParse.pl output. Alternatively, the utility fastx_clipper from the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) can be used instead of LibParse.pl to output de-multiplexed and adaptor-trimmed small RNA in FASTQ format for broader compatibility with downstream applications.
LibParse.pl example:
LibParse.pl -f in.fastq -t fastq -o libparse.prefix -r 50 -a libparse.failed -l libparse.log -v -e CTGTAG -E CGATGT,GATCAC,CAGATG,TACGTT,TTACCA,ACTGTA,ATCACG,ACTTGT,GCCAAT,TGCTAG,CTTGTA,TCAGGC
fastx_clipper example:
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATCGATGT -o out.I1.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATGATCAC -o out.I2.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATCAGATG -o out.I3.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATTACGTT -o out.I4.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATTTACCA -o out.I5.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATACTGTA -o out.I6.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATATCACG -o out.I7.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATACTTGT -o out.I8.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATGCCAAT -o out.I9.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATTGCTAG -o out.I10.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATCTTGTA -o out.I11.fastq
fastx_clipper -Q33 -l 18 -i in.fastq -c -v -M 23 -a CTGTAGGCACCATCAATTCAGGC -o out.I12.fastq
Recipes
- 37.5:1 acrylamide:bis solution for 7M-Urea, 17%-polyacrylamide gel and 6% native PAGE/0.5x TBE gel
300 g of acrylamide and 8 g of bisacrylamide dissolved in 300 ml pre-warmed deionized water. Once dissolved, adjust final volume to 1 L with warm deionized water. Filter sterilize with 0.45 µM bottle-top vacuum filter; wrap bottle in aluminum foil and stored at 4 °C.
- 10% ammonium persulfate (APS)
Add 10 g of ammonium persulfate to 80 ml of deionized water
Stir to dissolve
Adjust final volume to 100 ml
Syringe filter into 10 ml aliquots and store at -20 °C
- 2x RNA loading dye (50 ml)
47.5 ml of 100% formamide
125 µl of 10% SDS
0.0125 g of bromophenol blue
50 µl of 0.5 M EDTA
2.3 ml of H2O
Final concentration:
Formamide: 95%
SDS: 0.025%
Bromophenol blue: 0.025%
EDTA: 0.5 mM
- Low salt buffer (50 ml)
0.5 ml of 1 M Tris-HCl (pH 7.6)
1.0 ml of 5 M NaCl
0.1 ml of 0.5 M EDTA
48.4 ml of H2O
Final concentration:
Tris-HCl (pH 7.6): 10 mM
NaCl: 100 mM
EDTA: 1 mM
- High salt buffer (50 ml)
0.5 ml of 1 M Tris-HCl (pH 7.6)
10 ml of 5 M NaCl
0.1 ml of 0.5 M EDTA
0.526 g of L-Arginine
39.4 ml of H2O
Final concentration:
Tris-HCl (pH 7.6): 10 mM
NaCl: 1 M
EDTA: 1 mM
L-Arginine: 50 mM
Acknowledgments
The original version of this protocol was described in Carbonell et al. (2012). The updated version of the protocol was described in Carbonell et al. (2014). This work was supported by grants from the National Science Foundation (MCB-0956526, MCB-1231726) and National Institutes of Health (AI043288).
References
- Carbonell, A., Fahlgren, N., Garcia-Ruiz, H., Gilbert, K. B., Montgomery, T. A., Nguyen, T., Cuperus, J. T. and Carrington, J. C. (2012). Functional analysis of three Arabidopsis ARGONAUTES using slicer-defective mutants. Plant Cell 24(9): 3613-3629.
- Carbonell, A., Takeda, A., Fahlgren, N., Johnson, S. C., Cuperus, J. T. and Carrington, J. C. (2014). New generation of artificial MicroRNA and synthetic trans-acting small interfering RNA vectors for efficient gene silencing in Arabidopsis. Plant Physiol 165(1): 15-29.
- Dretzen, G., Bellard, M., Sassone-Corsi, P. and Chambon, P. (1981). A reliable method for the recovery of DNA fragments from agarose and acrylamide gels. Anal Biochem 112(2): 295-298.
- Munafo, D. B. and Robb, G. B. (2010). Optimization of enzymatic reaction conditions for generating representative pools of cDNA from small RNA. RNA 16(12): 2537-2552.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Gilbert, K. B., Fahlgren, N., Kasschau, K. D., Chapman, E. J., Carrington, J. C. and Carbonell, A. (2014). Preparation of Multiplexed Small RNA Libraries from Plants. Bio-protocol 4(21): e1275. DOI: 10.21769/BioProtoc.1275.
- Carbonell, A., Takeda, A., Fahlgren, N., Johnson, S. C., Cuperus, J. T. and Carrington, J. C. (2014). New generation of artificial MicroRNA and synthetic trans-acting small interfering RNA vectors for efficient gene silencing in Arabidopsis. Plant Physiol 165(1): 15-29.
Category
Plant Science > Plant molecular biology > RNA > RNA sequencing
Systems Biology > Transcriptomics > RNA-seq
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.