- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of T Cell Antigen-specific Memory Responses in Rhesus Macaques, Using Cytokine Flow Cytometry (CFC, also Known as ICS and ICCS): Analysis of Flow Data

Published: Vol 4, Iss 8, Apr 20, 2014 DOI: 10.21769/BioProtoc.1109 Views: 16760
Reviewed by: Jia LiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2012

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
What was initially termed ‘CFC’ (Cytokine Flow Cytometry) is now more commonly known as ‘ICS’ (Intra Cellular Staining), or less commonly as ‘ICCS’ (Intra Cellular Cytokine Staining). The key innovations were use of an effective permeant (allowing intracellular staining), and a reagent to disrupt secretion (trapping cytokines, thereby enabling accumulation of detectable intracellular signal). Because not all researchers who use the technique are interested in cytokines, the ‘ICS’ term has gained favor, though ‘CFC’ will be used here.
CFC is a test of cell function, exposing lymphocytes to antigen in culture, then measuring any cytokine responses elicited. Test cultures are processed so as to stain cells with monoclonal antibodies tagged with fluorescent markers, and to chemically fix the cells and decontaminate the samples, using paraformaldehyde.
CFC provides the powers of flow cytometry, which includes bulk sampling and multi-parametric cross-correlation, to the analysis of antigen-specific memory responses. A researcher using CFC is able to phenotypically characterize cells cultured with test antigen, and for phenotypic subsets (e.g. CD4+ or CD8+ T cells) determine the % frequency producing cytokine above background level.
In contrast to ELISPOT and Luminex methods, CFC can correlate production of multiple cytokines from particular, phenotypically-characterized cells. The CFC assay is useful for detecting that an individual has had an antigen exposure (as in population screenings), or for following the emergence and persistence of antigen memories (as in studies of vaccination, infections, or pathogenesis). In addition to quantifying the % frequency of antigen-responding cells, mean fluorescence intensity can be used to assess how much of a cytokine is generated within responding cells.
With the technological advance of flow cytometry, a current user of CFC often has access to 11 fluorescent channels (or even 18), making it possible to either highly-characterize the phenotypes of antigen-responding cells, or else simultaneously quantify the responses according to many cytokines or activation markers. Powerful software like FlowJo (TreeStar) and SPICE (NIAID) can be used to analyse the data, and to do sophisticated multivariate analysis of cytokine responses.
The method described here is customized for cells from Rhesus macaque monkeys, and the extensive annotating notes represent a decade of accumulated technical experience. The same scheme is readily applicable to other mammalian cells (e.g. human or mouse), though the exact antibody clones will differ according to host system. The basic method described here incubates 1 x 106 Lymphocytes in 1 ml tube culture with antigen and co-stimulatory antibodies in the presence of Brefeldin A, prior to staining and fixation.
Note: This is the second part of a two-part procedure. Part one has the same initial title, but the subtitle “From assay set-up to data acquisition (Sylwester et al., 2014)”. The Abstract and Historical Background is the same for both documents.
[Historical Background] In 1988, Andersson, et al. first demonstrated how lymphocytes could be fixed, permeabilized, stained with antibodies against IFNg, then fluorescently labeled and enumerated by flow cytometry. In 1991, Sander et al. demonstrated improved methods to fix cells with paraformaldehyde, permeabilize them with saponin, then use fluorescently-labeled antibodies to stain intracellular cytokines for microscopic examination. In 1993, Jung et al. extended this method to use with flow cytometry, and included the use of monensin to disrupt secretion, so as to increase intracellular signal of molecules otherwise released soon after synthesis. In 1995, Prussin and Metcalfe used directly-conjugated antibodies, and optimized the incubation period to 6 h. Also in 1995, Picker et al. considerably enhanced the sensitivity and reproducibility of cytokine detection by using Brefeldin A to block the secretion apparatus for cytokines, and by using a different permeant (Tween-20). In 1997, this matured method was applied by Picker et al. to study the the antigen-specific homeostatic mechanism in HIV+ patients. In 2001, Schuerwegh et al. confirmed that BfA provides for better cytokine signals than monensin, used by others in this method.
In two reports in 1989, one by Gardner et al. and the other by McClure et al. reported that Rhesus macaques were a useful model for studying HIV disease and AIDS. In 2002, Picker et al. reported the application of the CFC assay to Rhesus macaques. In 2012, a group created to develop multi-lab standards for use of ICS in NHP vaccine studies published their recommendations for a 96-well plate method with a 6 h total incubation (Donaldson et al., 2012 and Foulds et al., 2012).
The general procedure reported here is that 2002 tube-format method, now with a 9 h total incubation, and optimized especially for low-end sensitivity. The specific details here are the state of the art now practiced by the Louis Picker Lab, at the Oregon Health and Science University, affiliated with the Oregon National Primate Research Center. These methods have been used in several of our recent publications (Fukazawa et al., 2012; Hansen et al., 2011; Hansen et al., 2009). It is important to note that in our hands, plate-format ICS is not as reliable or sensitive for weak responses as is this tube-based method (unpublished observations). Until that problem is understood and solved, the tube-based method remains the most-sensitive format for CFC.
Materials and Reagents
- Data required: Flow cytometry data files, from CFC assay samples as described in “From Assay Set-up to Data Acquisition” (Sylwester et al., 2014).
Equipment
- Incubator for tissue culture (humidified, stable at 37 °C, 5% CO2 atmosphere)
- Flow cytometry analyser
Software
- Software-equipped workstation, for analysis of cytometry files
e.g. FlowJo (Tree Star)
e.g. SPICE (NIAID)
Procedure
- Choosing a gating strategy
Because there are so many ‘right’ ways to analyse data, this document will not attempt to impose a definitive prescription. Rather, this document presents one rigorous example that illustrates the role and value of advisable steps, some essential, others optional. This example is an illustration of the second of two mindsets one can have when gating: (1) An analyst can seek to minimize gating steps by custom-crafting well-fitting gates to each datafile. The advantages of this approach are fewer steps, and perhaps higher elegance. The drawbacks are that each gate will have many control points, and require careful and time-consuming adjustment when applied to a new file. This strategy is appealing when an analyst has relatively few files to gate, but cumbersome when confronted with hundreds or thousands of files. (2) Alternately, an analyst can seek to minimize time spent adjusting gates after applying a gating hierarchy to a set of files. This strategy makes use of many gates, each large and forgiving enough that they individually require little attention or adjustment as files change, but with the aggregate effect of ‘cleaning’, ‘focusing’, and ‘winnowing’ the data, and achieving the same quality of gating as the first strategy.
The illustration below is this second approach, making use of many ‘low-attention’ gates.- Outlined gating hierarchy
- ‘Data cleaning’ steps
- Data reduction gate (optional)
- Time vs CD45 (time advised, but CD45 optional)
- Singlets (advised)
- Drop aggregates (optional)
- CD45 vs CD3 (optional)
- Specific stain, CD3 cleanup (optional)
- Small lymphocytes (essential)
- Additional cleanup of T cell population (optional)
- Data reduction gate (optional)
- Division into CD4+ and CD8+ T cells
- CD4 vs CD8
- CD4+, CD4-, followed by CD8+ in CD4- population
- CD4 vs CD8
- Gating cytokine responses
- CD69 vs cytokine
- CD69 vs cytokine
- ‘Data cleaning’ steps
- Outlined gating hierarchy
- ‘Data cleaning’ steps
- Data reduction gate (optional)
Most analysis software depicts data as density plots, illustrated with topographical lines or color-coding. Often, the greatest event density is in the debris field in the lower left corner of a scatterplot, or in debris aggregates in the upper left corner. Since CFC is focused on analysis of lymphocytes, it is not necessary to leave in anything else. Unfortunately, when all these irrelevant events are in the scatterplot, their presence reduces the ‘resolution’ an analyst has regarding the margins of the lymphocyte population. Therefore, crudely removing most of the irrelevant data will improve resolution in the very next gate. It is possible to make a ‘Data Reduction’ gate that reliably works with all data files, needing only the most-minimal attention, yet significantly improving resolution immediately. An example is shown below: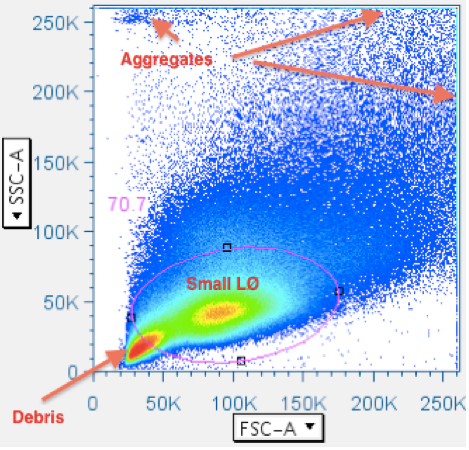
Notice that the highest event density is in the debris field in the lower left, and that the leftmost margin of the small lymphocyte population is obscured by blending with the rightmost margin of the debris field. The oval gate, in this example, removes 30% of the events from subsequent analysis. - Time vs CD45 (time advised, but CD45 optional; CD3 is an alternate option)
Cytometers that feed sample into the instrument by pressurizing commonly produce pressurizing artifacts when data recording begin and ends. Sometimes, other pressure effects (e.g., breakup of sample clumps) alter the data in between. It is therefore highly advised that the time-course data be checked, and data at the start/stop boundaries be removed. If the time gate data ‘centroid’ ever moves, it is likely that data placement has also shifted in downstream gates. It is therefore advisable that analysis only consider data segments with continuous time gate centroids.
It is not necessary to use CD45 for this analysis, but if CD45 can be used, it is convenient here. Otherwise, any parameter (CD3, or even SSC or FSC) is interchangeable with CD45 in the examples below.
Examples of ‘clean’ time data: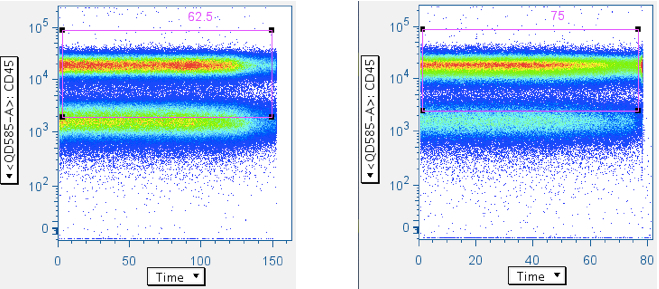
Note that the gate is generous along the CD45 axis. This is because we do not want to exclude CD45 dim WBC, and our general method is to apply many ‘low-maintenance’ gates, whose aggregate effect is to produce clean, focused data.
Examples of pressure artifacts not changing the data centroid: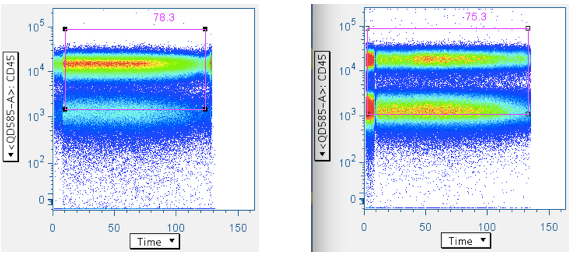
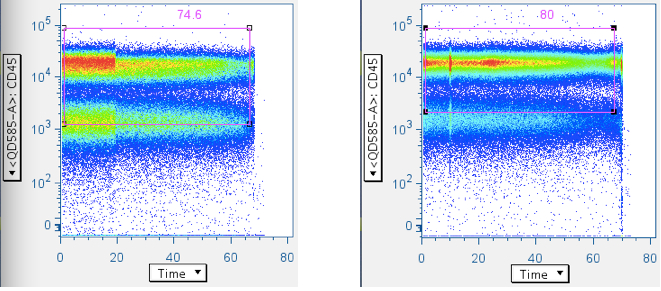
Example of a pressure effect changing the centroid, and affecting gating results downstream: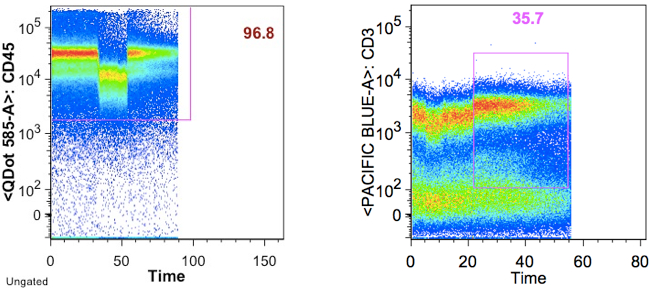
It is very important to exclude events that may be collected after a tube is sucked dry, since these events can affect cyotokine-response quantification.
Below is an example wherein the time gate extends past the sample, and the cytokine response plot that comes from it: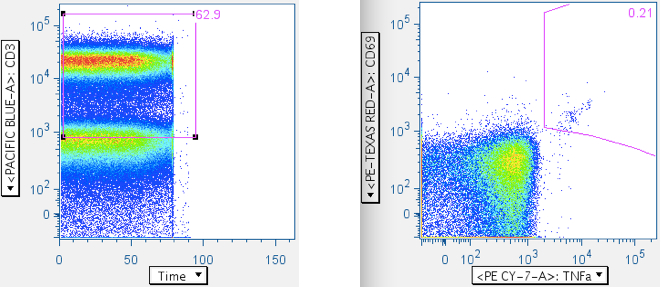
Here is the same datafile, with the time gate restricted to the sample. Notice the profound difference in the cyotokine response value, and the trivial cause of this difference.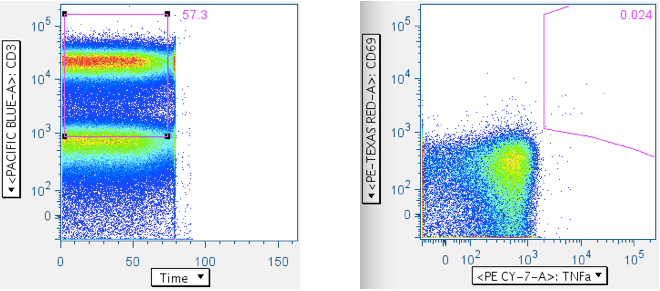
- Singlets (advised)
Amoeboid cells moving in a pipe tend to adopt a spherical shape, such that diameter is proportional to cross-sectional area. Therefore, if one plots FSC-H (H = height) vs FSC-A (A = area) (and/or SSC-H vs SSC-A), a line going up the midline should result. Deviations off that line can occur when two cells are stuck together (a ‘doublet’, forming a ‘figure-eight’). Triplets, quadruplets, and higher-order clusters look progressively like spheres again, so their deflections become less and less from the midline. But since the greatest deflection (doublets) is also the most common small cluster, it is easy to remove these contaminants (That might otherwise suggest that a particular event is simultaneously a B and T cell, or CD4+ and CD8+ cell, or might combine the response outcomes of two or three separate cells.). Other cytometrists prefer to plot FSC-H vs. FSC-W, because Area is an integrated value, whereas Height is a particle’s maximum signal and Width is a particle’s duration of signal (i.e., size), and independent of PMT voltage. What is described here is what the Picker Lab actually does. If Area Scaling is set properly (with cells), H vs A should line up at the midline. If two (or more) cells are stuck together, signal area will no longer line up with signal height.
Examples of this gate is given below: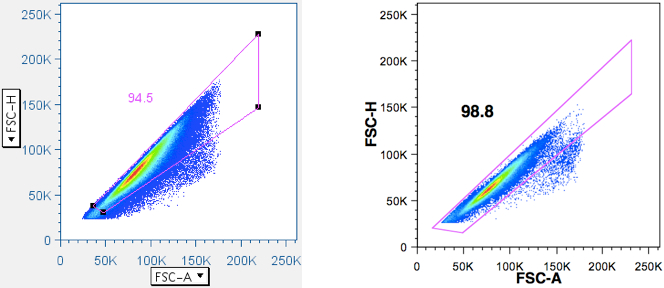
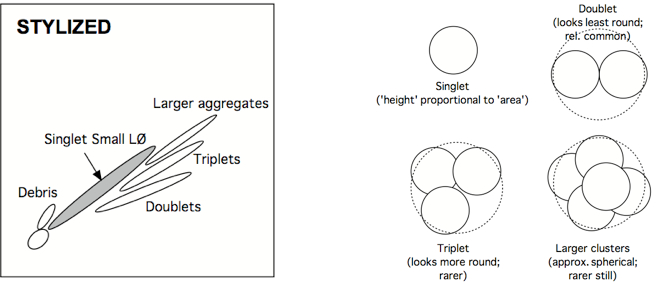
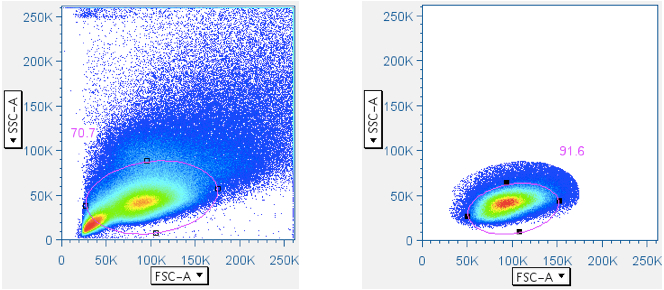
Note: The debris in the lower left corner of the displayed data. In the first example it is gated out. In the second example, the gate could be improved by adjusting it to reduce that debris. The subpopulations are shown more clearly in the stylized illustration. - Drop aggregates (optional)
Although the singlet gate removes most of the low-cell clusters from analysis, the singlet gate does poorly with higher-order clusters that made it past the ‘data reduction’ gate. Even when these are rare, they can cause havoc when a researcher is focusing on very weak responses, because even a few such events can significantly affect the % in a gate of very few events. Fortunately, it is simple to install a low-maintenance gate that reliably removes the threat of these events, a gate that takes advantage of the fact that clusters of cells will behave as events (‘cells’) that are unnaturally bright.
Be careful with this gate, however, because you can sometimes get very robust responses like:
In the five panels below, it is important to notice that these axes are logarithmic, but include zero, and have events apparently extending below ‘zero’, and off the plot. When crafting any gate meant to include events below the lower limit of the axis, be sure to create an oversized gate that extends WELL past the lower boundary. That way, it is less likely you’ll unintentionally truncate your data. In the fifth plot below, note that the cytokine-positive events are so bright that they’re going off-scale. In this case, it is important to extend the gate off-scale so that these events are captured. The drawback to this is that aggregates will also be perpetuated in the data.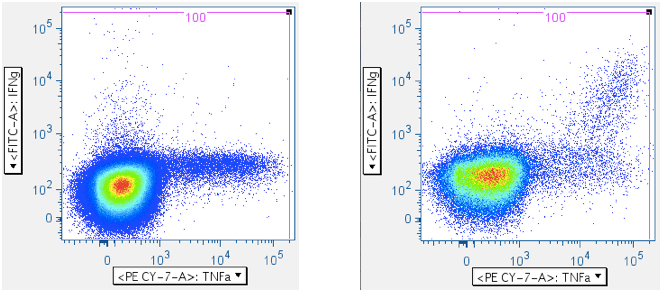
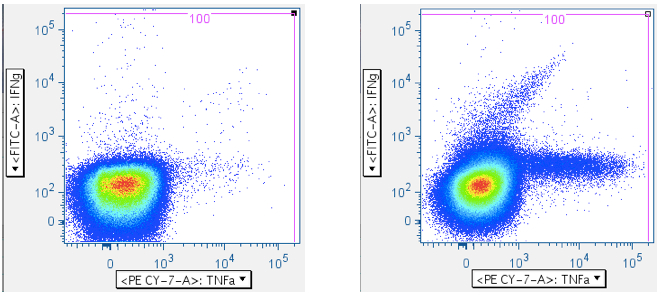
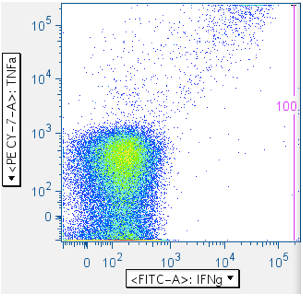
In the examples above, the first panel is a no-antigen (no-response) sample, still with considerable pre-gated TNFa signal, and with some IFNg signal. The second panel of the upper row shows cells responding to superantigen SEB, with a notable population that is responding with both IFNg and TNFa. The first panel of the second row shows a population weakly responding to SIV Gag peptide mix. The last panel shows a diagonal artifact commonly induced by toxic peptides in SIV Env peptide mixes. Our lab refers to this as the ‘death spike’, and whichever of these events that persist through gating contaminate downstream plots by running up the midline. Leaving this artifact in can dramatically – and incorrectly -- inflate the reported response.
Note: In all the examples above, there are super-bright events on the top and far-right axes. - CD45 vs CD3 (optional)
If the staining panel has incorporated CD45, an excellent use is to apply it here. CD45 is a marker on all leukocytes (WBC), and so this particular gate is highly valuable when the CFC is investigating responses in tissues in which lymphocytes are a minority population [e.g. lung wash (< 5%), small intestine, liver, vaginal mucosa, etc.]. Without CD45, one can still reduce non-leukocyte contamination with a CD3 vs CD4 (or CD8) gate, but it doesn’t produce as satisfying as cleanse. Moreover, this gate allows some reduction of any ‘death spike’ that’s present, or subcellular debris released when cells go through a freeze-thaw cycle.
Examples of this gate are shown below: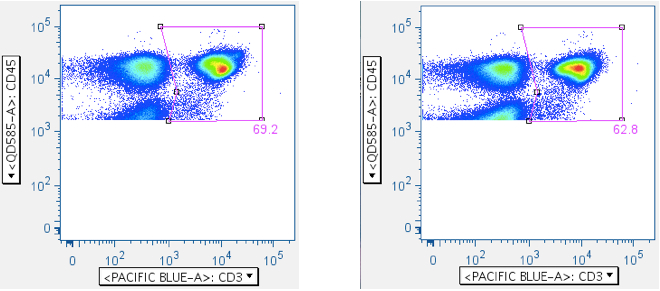
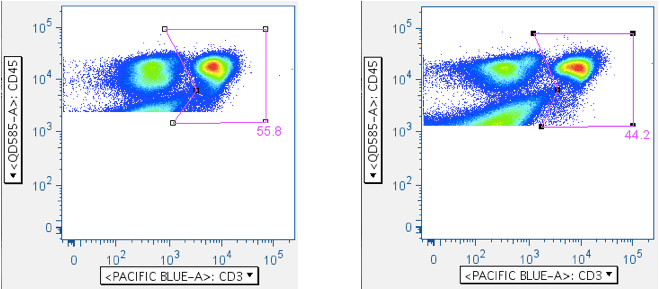
The top row shows a negative and weakly-responding sample, respectively. The bottom row shows samples showing the ‘death spike’ induced by SIV Env toxic peptides. Notice how this gate can reduce death spike events from downstream analysis.
In the absence of a death spike, this gate tends to be reliably low-maintenance, but very effective at cleaning out debris that is otherwise hard to remove. - Specific stain, and CD3 cleanup 1 (optional)
If your panel allows the luxury of an unused channel, you can obtain benefit from actually collecting data from that parameter. The idea is simple: If you have positive events in an unused channel, they cannot possibly be valid. Therefore, this gate is an opportunity to remove them, and it is very valuable when trying to measure very weak, threshold-level responses.
But this same gate can serve another purpose when the antigen roster includes something like SIV Env peptide mix, which includes toxic peptides. It can be very difficult to remove all the cells affected by these toxic peptides, because live-dead stains don’t always gate them out, and because the events characteristic of a ‘death spike’ run up the midline, often obscuring populations that interest us. By using a plot that combines an unused channel vs CD3, the CD3+ population that interests us is shoved off the midline, and any death spike ‘glances’ it, in a way that can be partially gated out.
Examples are shown below:
The first panel shows a typical, well-behaved sample. The second panel in the top row shows the location of Env toxic peptide-induced carcasses (notice how the gate reduces them from the downstream data). The second row panel shows an example with a surprising amount of positive signalling, even though that fluorophore was not used in the stain.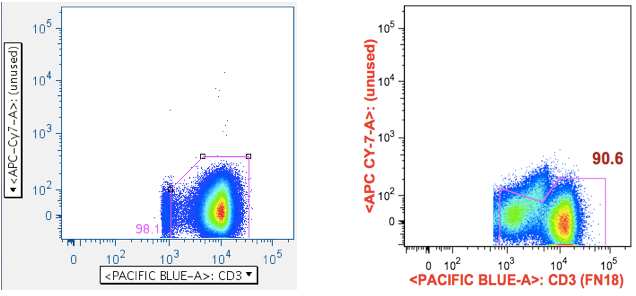
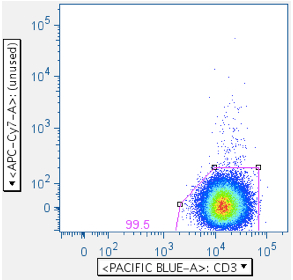
- Small lymphocytes (essential)
In CFC, it is essential that the analysis be focused first, in a scatterplot, on lymphocytes, then separately by CD3 fluorescence, on the subset of small lymphocytes that are T cells. Thus, this gate is essential. In the gating scheme shown here, the first gate (‘Data Reduction’) crudely approximated the small lymphocyte population. But that gate is often blended with the debris field, and in some tissues (e.g. lung wash, intestine, liver) the debris can be so overwhelming that an analyst is forced to guess where the lymphocytes are in a scatterplot. Moreover, it is important to stress that pure B lymphocytes and NK lymphocytes have scatter profiles distinct, though overlapping, with T lymphocytes. It is therefore beneficial to apply the ‘Small Lymphocyte’ gate after the population has gone through a CD3+ gate.
Below are examples of original scatterplots, showing how well the previous steps have clarified the data and improved the resolution of the lymphocytes, and how a tighter (and more believable) ‘Small Lymphocyte’ gate can now be applied: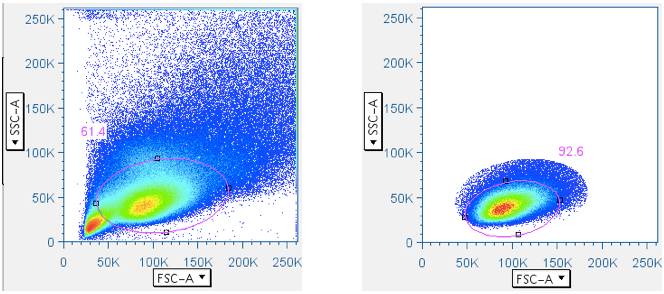
- Additional cleanup to the T cell (CD3+ small lymphocytes) population (optional)
Discrete populations often have a ‘fuzz’ of events between them, complicating the decision of where to place gates. Some plots make the decision clearer, and this gating step is like that. At this stage, we have a fairly clean population of T cells, except for the problem that the bright CD3+ is usually trailed by a smear of CD3-medium events, down into a CD3-population (which we have removed). The problem is that CD3-medium population; should we (can we) gate any parts out?
By plotting CD3 vs CD69 (an activation marker), or alternately by plotting CD3 vs a cytokine, the CD3-medium population is resolved in a way that helps with gating: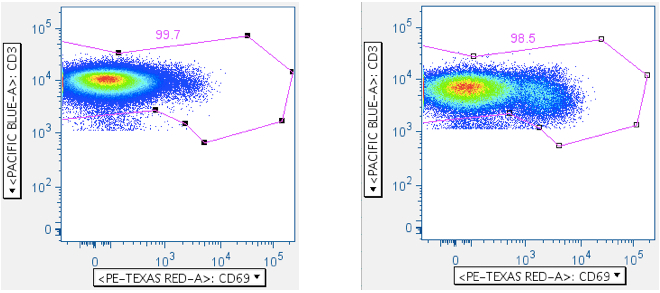
The first panel above shows a sample lacking any test antigen; the second panel shows sample stimulated with superantigen SEB. Note how in both cases it’s clear where to draw the gate against CD3-medium/dim cells.
Below, a different SEB sample is shown first by CD3 vs CD69, and then by CD3 vs TNFa. Either way can be used, though CD69 activation is more reliable than TNFa, since not all cells elaborating TNFa also elaborate IFNg or IL2, etc.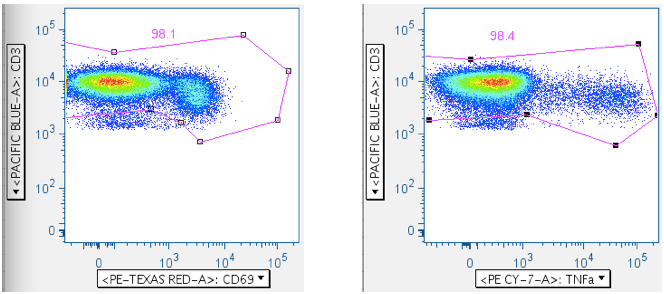
This gate is also useful for removing more ‘death spike’ cells from analysis, as the SIV Env peptide mix examples below illustrate: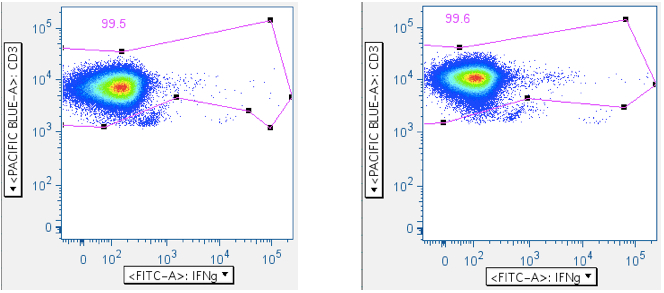
- Data reduction gate (optional)
- Division into CD4+ and CD8+ T cell populations (essential)
Now that one has ‘clean’ T cells, there are two ways of separating them into separate CD4+ and CD8+ T cell bins: (1) A direct plot of CD4 vs CD8. This has the virtue of simplicity, but the drawback of sometimes having ambiguity with where to put the CD4+CD8+ double-positive population (If, indeed, it will be included at all; in the examples below, the CD4+CD8+ population is included with the CD4+ T cells. A user can opt to gate the CD4+CD8+ double-positive T cells as a separate, unique population.). (2) A two-step approach that yields more consistent results, but involves more input.- CD4 vs CD8
The direct approach: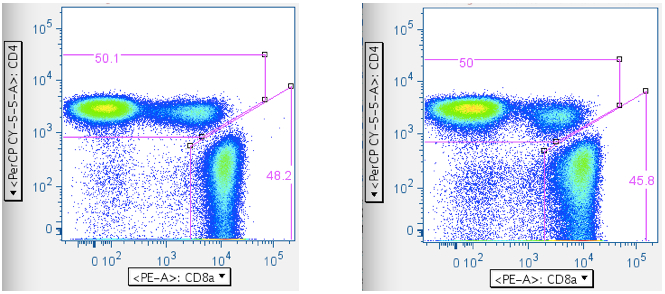
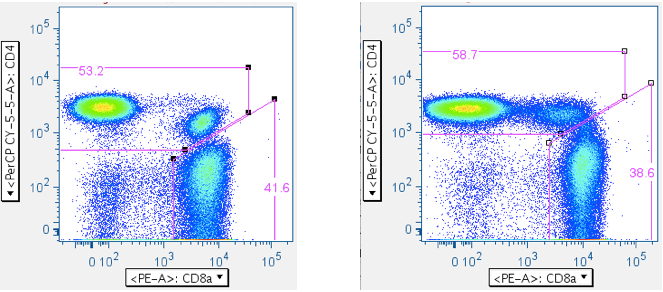
- Reducing the SIV Env toxic peptide-induced ‘death spike’:
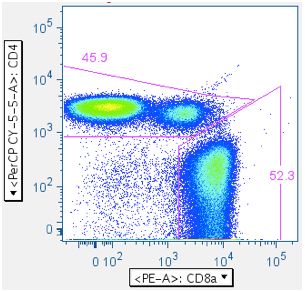
- (CD4+, CD4-), followed by (CD8+ in CD4- population)
The two-step approach: (1) CD4+, CD4-, then (2) regate CD4- for CD8+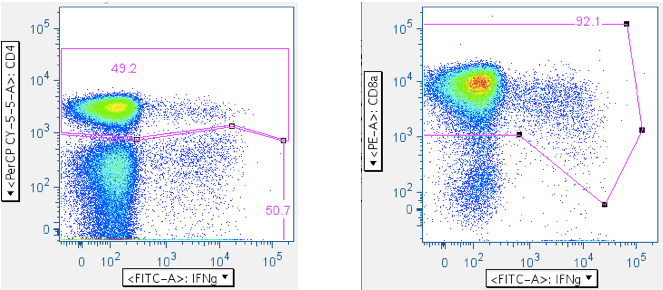
Reducing the SIV Env toxic peptide ‘death spike’: The goal is to remove the debris spike, and the gate-shape above is just a suggestion.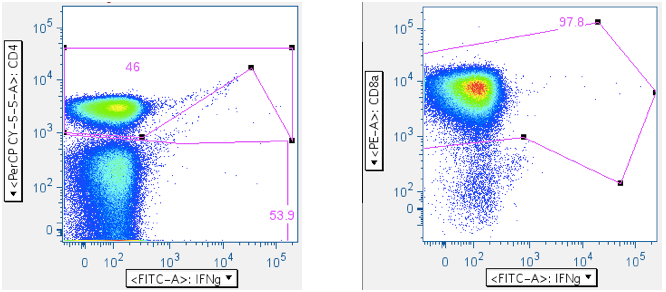
- CD4 vs CD8
- Gating cytokine responses
- CD69 vs cytokine
Everything up to this point has been with the goal of getting maximally-clean populations of CD4+ and CD8+ T cells. The last step is to gate the antigen-stimulated cytokine-positive cells. The Picker Lab plots activation marker CD69 vs the various cytokines (TNFa, IFNg, MIP1b, IL2, etc), because we have seen sometimes significant cytokine signal emanating from unactivated (i.e., CD69-) cells. If your tests produce robust positive responses, this may not matter to you. However, if you, like us, are interested in very weak responses, then inclusion of any events that aren’t actually antigen-stimulated is a degradation of assay sensitivity.
Example of a ‘negative’ sample, showing IL2 emanating from the CD69- population: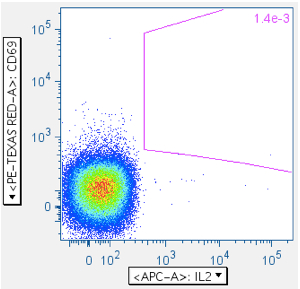
Examples of how SEB responses are gated.
Note: The numbers in the upper right corners report the % of all events in the plot that are in the gate; that is, the ‘% frequency of responding cells.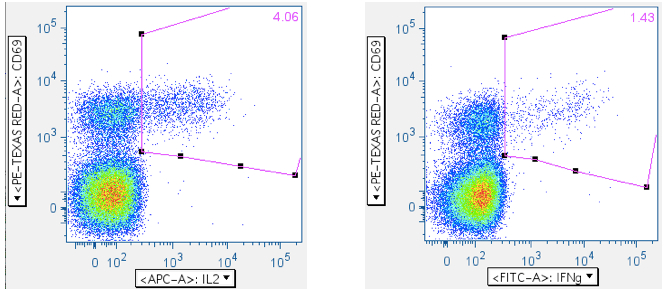
Example of an SIV Gag peptide mix response: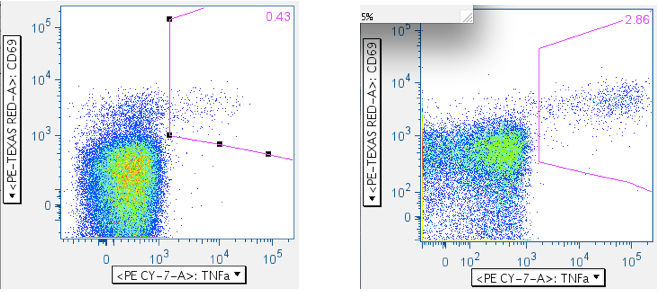
- CD69 vs cytokine
Acknowledgments
The methods described in this protocol have been evolving since the 1995 paper cited in this protocol (Picker et al., 1995), and were used in the two 2013 papers published by our group (Hansen et al., 2013a and Hansen et al., 2013b). Funding has been provided by NIH and by the Bill and Melinda Gates Foundation.
References
- Andersson, U., Hallden, G., Persson, U., Hed, J., Moller, G. and DeLey, M. (1988). Enumeration of IFN-γ-producing cells by flow cytometry. Comparison with fluorescence microscopy. J Immunol Methods 112(1): 139-142.
- Donaldson, M. M., Kao, S. F., Eslamizar, L., Gee, C., Koopman, G., Lifton, M., Schmitz, J. E., Sylwester, A. W., Wilson, A., Hawkins, N., Self, S. G., Roederer, M. and Foulds, K. E. (2012). Optimization and qualification of an 8-color intracellular cytokine staining assay for quantifying T cell responses in rhesus macaques for pre-clinical vaccine studies. J Immunol Methods 386(1-2): 10-21.
- Foulds, K. E., Donaldson, M. and Roederer, M. (2012). OMIP-005: Quality and phenotype of antigen-responsive rhesus macaque T cells. Cytometry A 81(5): 360-361.
- Fukazawa, Y., Park, H., Cameron, M. J., Lefebvre, F., Lum, R., Coombes, N., Mahyari, E., Hagen, S. I., Bae, J. Y., Reyes, M. D., 3rd, Swanson, T., Legasse, A. W., Sylwester, A., Hansen, S. G., Smith, A. T., Stafova, P., Shoemaker, R., Li, Y., Oswald, K., Axthelm, M. K., McDermott, A., Ferrari, G., Montefiori, D. C., Edlefsen, P. T., Piatak, M., Jr., Lifson, J. D., Sekaly, R. P. and Picker, L. J. (2012). Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat Med 18(11): 1673-1681.
- Gardner, M. B. (1989). SIV infected rhesus macaques: an AIDS model for immunoprevention and immunotherapy. Adv Exp Med Biol 251: 279-293.
- Hansen, S. G., Vieville, C., Whizin, N., Coyne-Johnson, L., Siess, D. C., Drummond, D. D., Legasse, A. W., Axthelm, M. K., Oswald, K., Trubey, C. M., Piatak, M., Jr., Lifson, J. D., Nelson, J. A., Jarvis, M. A. and Picker, L. J. (2009). Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 15(3): 293-299.
- Hansen, S. G., Ford, J. C., Lewis, M. S., Ventura, A. B., Hughes, C. M., Coyne-Johnson, L., Whizin, N., Oswald, K., Shoemaker, R., Swanson, T., Legasse, A. W., Chiuchiolo, M. J., Parks, C. L., Axthelm, M. K., Nelson, J. A., Jarvis, M. A., Piatak, M., Jr., Lifson, J. D. and Picker, L. J. (2011). Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473(7348): 523-527.
- Hansen, S. G., Sacha, J. B., Hughes, C. M., Ford, J. C., Burwitz, B. J., Scholz, I., Gilbride, R. M., Lewis, M. S., Gilliam, A. N., Ventura, A. B., Malouli, D., Xu, G., Richards, R., Whizin, N., Reed, J. S., Hammond, K. B., Fischer, M., Turner, J. M., Legasse, A. W., Axthelm, M. K., Edlefsen, P. T., Nelson, J. A., Lifson, J. D., Fruh, K. and Picker, L. J. (2013). Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340(6135): 1237874.
- Hansen, S. G., Piatak, M., Jr., Ventura, A. B., Hughes, C. M., Gilbride, R. M., Ford, J. C., Oswald, K., Shoemaker, R., Li, Y., Lewis, M. S., Gilliam, A. N., Xu, G., Whizin, N., Burwitz, B. J., Planer, S. L., Turner, J. M., Legasse, A. W., Axthelm, M. K., Nelson, J. A., Fruh, K., Sacha, J. B., Estes, J. D., Keele, B. F., Edlefsen, P. T., Lifson, J. D. and Picker, L. J. (2013). Immune clearance of highly pathogenic SIV infection. Nature 502(7469): 100-104.
- Jung, T., Schauer, U., Heusser, C., Neumann, C. and Rieger, C. (1993). Detection of intracellular cytokines by flow cytometry. J Immunol Methods 159(1-2): 197-207.
- McClure, H. M., Anderson, D. C., Fultz, P. N., Ansari, A. A., Lockwood, E. and Brodie, A. (1989). Spectrum of disease in macaque monkeys chronically infected with SIV/SMM. Vet Immunol Immunopathol 21(1): 13-24.
- Picker, L. J., Singh, M. K., Zdraveski, Z., Treer, J. R., Waldrop, S. L., Bergstresser, P. R. and Maino, V. C. (1995). Direct demonstration of cytokine synthesis heterogeneity among human memory/effector T cells by flow cytometry. Blood 86(4): 1408-1419.
- Pitcher, C. J., Hagen, S. I., Walker, J. M., Lum, R., Mitchell, B. L., Maino, V. C., Axthelm, M. K. and Picker, L. J. (2002). Development and homeostasis of T cell memory in rhesus macaque. J Immunol 168(1): 29-43.
- Prussin, C. and Metcalfe, D. D. (1995). Detection of intracytoplasmic cytokine using flow cytometry and directly conjugated anti-cytokine antibodies. J Immunol Methods 188(1): 117-128.
- Sander, B., Andersson, J. and Andersson, U. (1991). Assessment of cytokines by immunofluorescence and the paraformaldehyde-saponin procedure. Immunol Rev 119: 65-93.
- Schuerwegh, A. J., Stevens, W. J., Bridts, C. H. and De Clerck, L. S. (2001). Evaluation of monensin and brefeldin A for flow cytometric determination of interleukin-1β, interleukin-6, and tumor necrosis factor-alpha in monocytes. Cytometry 46(3): 172-176.
- Sylwester, A. W., Scott, G. H. and Louis, J. P. (2014). Quantification of T cell antigen-specific memory responses in rhesus macaques, using cytokine flow cytometry (CFC, also known as ICS and ICCS): from assays et-up to data acquisition. Bio-protocol 4(8): e1110.
- Waldrop, S. L., Pitcher, C. J., Peterson, D. M., Maino, V. C. and Picker, L. J. (1997). Determination of antigen-specific memory/effector CD4+ T cell frequencies by flow cytometry: evidence for a novel, antigen-specific homeostatic mechanism in HIV-associated immunodeficiency. J Clin Invest 99(7): 1739-1750.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sylwester, A. W., Hansen, S. G. and Picker, L. J. (2014). Quantification of T Cell Antigen-specific Memory Responses in Rhesus Macaques, Using Cytokine Flow Cytometry (CFC, also Known as ICS and ICCS): Analysis of Flow Data. Bio-protocol 4(8): e1109. DOI: 10.21769/BioProtoc.1109.
Category
Immunology > Immune cell function > Antigen-specific response
Immunology > Immune cell staining > Flow cytometry
Cell Biology > Cell-based analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.