- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Determination of the Intracellular Calcium Concentration in Peritoneal Macrophages Using Microfluorimetry
Published: Vol 3, Iss 23, Dec 5, 2013 DOI: 10.21769/BioProtoc.988 Views: 18131
Reviewed by: Fanglian He
Original research article
The authors used this protocol in:

Apr 2013

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Calcium is one of the most important intracellular messengers in biological systems. Ca2+ microfluorimetry is a valuable tool to assess information about mechanisms involved in the regulation of intracellular Ca2+ levels in research on cells and in living tissues. In essence, the use of a dye that fluoresces in the presence of a target substance allows the detection of changes in the concentration of this molecule by determining the changes in the fluorescence of the probe (increases or decreases, depending on the nature of the dye used; for a review see Tsien et al. 1985). In this regard, there have been developed two different methodologies to assess intracellular Ca2+ measurements. On the one hand, ratiometric methods are based on the use of a ratio between two fluorescence intensities linked to the physicochemical properties of the probe. This allows correction of artifacts due to bleaching, changes in focus, variations in laser intensity, etc. but makes measurements and data processing more complicated since they require more expensive equipment with the possibility to change the wavelength emission/detection in a rapid way. Some ratiometric Ca2+ indicators are Fura-2 and Indo-1. On the other hand, on binding to Ca2+, indicators used for non-ratiometric measurements show a shift in their fluorescence intensity (the free indicator has usually a very weak fluorescence). Therefore, although an increase in fluorescence signal can be related directly to an increase in Ca2+ concentration, the fluorescence intensity depends on many factors such as acquisition conditions, probe concentration, optical path length, balance between the affinity constants of proteins binding Ca2+, among others. However, the fluxes of Ca2+ are of such a magnitude that these interferences are minor contributors to biases in the measurements. There are many non-ratiometric calcium indicators, some of which are Fluo-3, Fluo-4 and Calcium-Green-3. Consequently, the most suitable Ca2+-probe for each experiment will depend on the range of Ca2+ concentration that has to be evaluated, instrumentation, loading requirements, etc. In the present report we describe the protocol employed to quantify intracellular Ca2+ changes in peritoneal macrophages using Fura-2 as a fluorimetric probe and a microfluorimetric protocol that allows quantification of responding cells to a given stimulus, localization of the main intracellular domains sensing Ca2+ changes and a time-resolved analysis of the Fura-2 fluorescence that reflects the intracellular dynamics of Ca2+ in these cells (Través et al., 2013).
Keywords: Calcium fluxesMaterials and Reagents
- Fura 2-AM (Life Technologies)
- DMSO (Sigma-Aldrich)
- EGTA
- CaCl2
- HEPES
- Trypan Blue
- FCS
- Vacuum grease
- Peritoneal macrophages obtained from Balb/c male mice (8-12 weeks old) 4 days after i.p. administration of 2.5 ml of 3% thioglycollate solution
Note: In our study (Traves et al., 2013), the effect of prostaglandin E2 on the response to P2X/P2Y agonists ATP and BzATP was studied in depth. All these molecules were purchased from Sigma-Aldrich (St. Louis, MO). - 10x DPBS without Ca2+ Mg2+ (Lonza)
- Locke's solution (see Recipes) (supplemented with 1 mg/ml BSA)
Equipment
- Coverslips 0.13-0.16 mm thickness (15 mm of diameter) (SCHOTT AG)
- Forceps
- 35 mm culture dishes
- Basic water-jacket CO2 incubator
- Perfusion chamber RC-25F (Warner Instruments)
- Valves employed for cell perfusion (Warner Instruments)
- Perfusion valves controller VC 8 (Warner Instruments)
- Vacuum pump
- Water bath
- NIKON TE-200 microscope with a Plan Fluor x20/0.5 water objective( Nikon Corporation, model:TE-200)
- Dichroic mirror (430 nm) and a 510 nm band-pass filter (Omega Optical)
- ORCA-ER C4742-80 camera (Hamamatsu Photonics K. K., model: C4742-80)
- Filter wheel Lambda 10-2 (Sutter Instrument Company)
Software
- MetaFluor 6.2r & PC software (UNIVERSAL SOLUTIONS)
Procedure
- Firstly, a sterile coverslip was placed in the bottom of the culture dish (forceps may be helpful) where cells stuck from the very beginning. Macrophages were then carefully seeded in 1 ml of supplemented Locke's solution at a density of 300,000- 500,000 cells per 35 mm culture dish. Since they are non-dividing cells they were used in the following two or three days after seeded (likely, after this lapse time, macrophages will be completely adhered to coverslips). Extensive washing with 500 μl PBS 1X (three-four replicates) was used to ensure that cells were very adherent and alive. The percentage of apoptotic/necrotic cells after 3 days in culture was below 5% (determined by using the common Trypan Blue staining, see Notes).
Note: Cell attaching to the coverslip is a critical step in order to perform the microfluorimetric analysis. If your plastic or glass interferes with the adherence of the macrophages, allow the support to dry with FCS (fetal calf serum) under the cabin to improve adherence. Avoid a high density cell culture. A 50-75% subconfluent culture with some cells interacting is desirable. - Incubate macrophages in 1 ml supplemented Locke's solution loaded with 5-7 μM Fura-2-AM (dissolved in DMSO, following the datasheet instructions) for 45 min approximately at 37 °C. Incubation does NOT involve either shaking or agitation, just a short and slightly balancing.
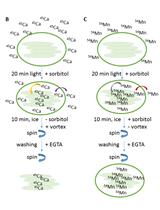
- Wash thoroughly the culture monolayer with fresh 500 μl Locke's solution (serum also contains esterases that may degrade Fura-2-AM) and place the coverslip in a small superfusion chamber (34 μl volume). Junctions between the coverslip and the chamber are sealed with vacuum grease. Figure 1 depicts the superfusion chamber apparatus.
Note: Locke’s media or products perfused to the chamber are regulated using a valve system that works by gravity. The perfusion solutions are maintained in a water bath at 37 °C and the flow rate is kept constant at 1.5 ml/min. The vacuum pump aspires continuously the perfusion media after arriving to the chamber to prevent the accumulation of the hydrolysis products.
Figure 2 shows the experimental setup described here.
Figure 1. The superfusion chamber system. In the upper side of the figure, we can appreciate the superfusion chamber and the microscope stage. Immediately below these two pictures, both pieces are already assembled. The coverslip should be placed in the circular hollow remaining in the center.
Figure 2. Experimental equipment for Fura-2-AM registers. The temperature and the valve controllers, the fluorescence microscope (including its optical filter changer) and the necessary equipment to record the fluorescence images (the camera and its controller) are depicted above. - Images of both control and treated cells are visualized using the Plan Fluor x20/0.5 objective of the microscope. In this regard, Fura-2-AM has entered the cell and intracellular esterases have hydrolyzed the compound providing free Fura-2 that senses Ca2+ with a high affinity. Fluorescence emission occurs in a broad range around 430 nm when the cells are excited at 340 nm.
Note: In our study (Traves et al., 2013), macrophages were preincubated with different prostanoids for at least 10 min and then stimulated for 30 s with a variety of purinergic receptor agonists at near-maximal effective concentrations: 100 μM ATP, 100 μM UTP, 10 μM UDP, 10 μM 2MeSADP, 1,000 μM α,β-meATP or 300 μM BzATP. - Excite cells for 300 ms at 340/380 nm (< 5 ms wavelength change) and select the emitted light using the dichroic mirror (430 nm) and a 510-nm band-pass filter.
Note: The selection of these wavelengths matches with the maximum fluorescence registers for Fura-2 calcium-saturated solutions (340 nm) and calcium-free Fura-2 solutions (380 nm). - Fluorescence images are acquired with the camera every 1.5 seconds and controlled by the software. Sampling frequency is 2 Hz.
Data analysis
- Images are processed by averaging signals from small elliptical regions within individual cells (Figure 3). The possibility exists to define specific areas of changes in the fluorescence emission (cell contact interactions, protrusions in the cytoplasm –cell polarization- or simply cells with different morphologies –round vs. shaped macrophages, depending on the treatments-).
Note: Background signals are subtracted from each wavelength.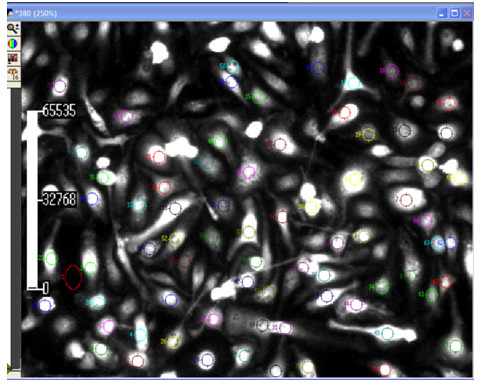
Figure 3. Metafluor screenshot of cells. ROIs (cells in which a shift in the fluorescence emission is recorded as a function of time) and background (normally areas without cells or non-responsive cells during the period of observation) are marked with a dot in the image. The scale bar on the left side refers to the time variable (in seconds). - The F340/F380 ratio is calculated on the basis of the initial peak magnitude that represents the initial transient components (Figure 4). The F340/F380 ratio is converted into a known calcium concentration using the Grynkiewicz equation:
[Ca2+] = Kd * (R – Rmin) / (Rmax – R) * F380max/F380min (Grynkiewicz et al., 1985)
Where:- Kd is the dissociation constant (depends on the indicator, but also on pH, ionic strength, cell line, etc.).
- R is the observed fluorescence ratio at both wavelengths (F340/F380).
- Rmin is the minimum ratio value (in absence of Ca2+).
- Rmax is the maximum ratio value (when Fura-2 is saturated by Ca2+).
- F380max/F380min is a scaling factor (fluorescence intensity at 380 nm excitation in the absence of Ca2+ and at Ca2+ saturation).
Note: A calibration curve is required to calculate the Kd value. The F380max and F380min values are obtained at the end of each analysis in the same experimental conditions (Fura-2-AM concentration, exposition time…). It is based in two calibration points, a maximum and a minimum corresponding, respectively, to a saturated calcium solution (2.5 mM CaCl2) and a calcium-free solution (containing 10 μM EGTA). Alternatively, inhibition of the membrane reticulum Ca2+ pump with 200 nM thapsigargin or 500 nM ter-buthylbenzohydroquinone allow a saturation of the cytoplasmic Ca2+ concentration. Treatment of cells maintained in extracellular medium lacking or containing Ca2+ with a low dose of ionomycin (ca. 1 μM) to improve the entrance of the Ca2+ into the cell.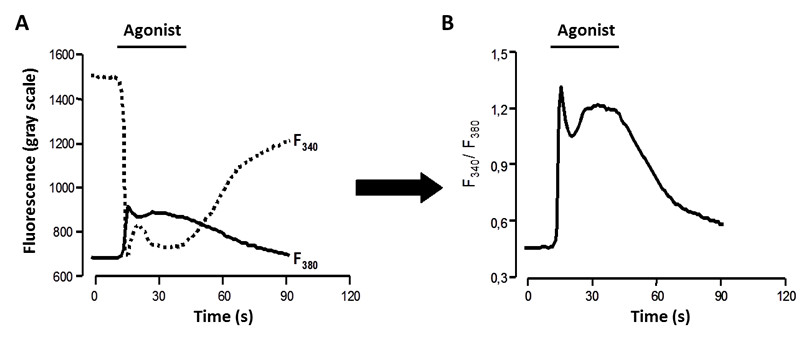
Figure 4. Fluorescence measured at 340/380 nm. A. Fluorescence intensity at both registered excitation wavelengths. B. Registers obtained over the time are divided to determine the F340/F380 ratio.
- Kd is the dissociation constant (depends on the indicator, but also on pH, ionic strength, cell line, etc.).
Notes
- Trypan Blue staining can be used to discriminate between viable and non-viable cells. The protocol follows these typical steps:
- Dilute the cell sample (1:10) to a total volume of 20 μl in a 0.4% Trypan Blue dye solution (should be sterile filtered before using).
- While non-viable cells will be blue, viable cells will be unstained.
- Carefully and continuously fill the hemocytometer chamber with 10 μl of the solution each chamber (all hemocytometers consist of two chambers; each is divided into nine 1mm2 squares).
- Count cells under the microscope in four 1 x 1 mm squares of one chamber and determine the average number of cells per square. If the cell density is higher than 200 cells/square, you should dilute your cell suspension.
- Total number of particles per ml in the cell sample can be calculated as follows: mean number of cells x 1/dilution factor x 104 cells/ml.
- Dilute the cell sample (1:10) to a total volume of 20 μl in a 0.4% Trypan Blue dye solution (should be sterile filtered before using).
Recipes
- Locke's Solution composition
140 mM NaCl
4.7 mM KCl
2.5 mM CaCl2
1.2 mM KH2PO4
1.2 mM MgSO4
5.5 mM glucose
10 mM HEPES, pH 7.4
Acknowledgments
This work was supported by grants CP11/00080 from ISCIII, BFU2011-024760 from MICINN and FIS-RECAVA RD12/0042/0019. RECAVA and Ciberehd networks are funded by the Carlos III Health Institute. A summary of the procedure was described in Traves et al. (2013).
References
- Traves, P. G., Pimentel-Santillana, M., Carrasquero, L. M., Perez-Sen, R., Delicado, E. G., Luque, A., Izquierdo, M., Martin-Sanz, P., Miras-Portugal, M. T. and Bosca, L. (2013). Selective impairment of P2Y signaling by prostaglandin E2 in macrophages: implications for Ca2+-dependent responses. J Immunol 190(8): 4226-4235.
- Grynkiewicz, G., Poenie, M. and Tsien, R. Y. (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260(6): 3440-3450.
- Traves, P. G., Pimentel-Santillana, M., Carrasquero, L. M., Perez-Sen, R., Delicado, E. G., Luque, A., Izquierdo, M., Martin-Sanz, P., Miras-Portugal, M. T. and Bosca, L. (2013). Selective impairment of P2Y signaling by prostaglandin E2 in macrophages: implications for Ca2+-dependent responses. J Immunol 190(8): 4226-4235.
- Tsien, R. Y., Rink, T. J. and Poenie, M. (1985). Measurement of cytosolic free Ca2+ in individual small cells using fluorescence microscopy with dual excitation wavelengths. Cell Calcium 6(1-2): 145-157.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
González-Ramos, S., Carrasquero, L. M. G., Delicado, E. G., Miras-Portugal, M. T., Fernández-Velasco, M. and Boscá, L. (2013). Determination of the Intracellular Calcium Concentration in Peritoneal Macrophages Using Microfluorimetry. Bio-protocol 3(23): e988. DOI: 10.21769/BioProtoc.988.
Category
Immunology > Immune cell function > Macrophage
Biochemistry > Other compound > Ion > Calcium
Cell Biology > Cell imaging > Microfluidics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.