- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

[3H]-Spiperone Competition Binding to Dopamine D2, D3 and D4 Receptors
Published: Vol 3, Iss 20, Oct 20, 2013 DOI: 10.21769/BioProtoc.944 Views: 9705
Reviewed by: Fanglian HeCheng ZhangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2012
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
This protocol is intended for use in 96 well plates (1,200 μl wells) but it can similarly be applied to standard test tubes (Levant, 2007). D2, D3, and D4 dopamine receptors are members of the D2-like class of dopamine receptors. They can be studied using the radioligand [3H]-spiperone, which is an antagonist binding to D2, D3, and D4 receptors with comparable affinity. A competition experiment is usually performed to determine the affinity of a compound for a receptor. If multiple subtypes or states of the receptor are present and the competing compound differentiates them, a competition binding experiment can quantify the relative contribution of the two subtypes or states; while resolution of more than two subtypes or states is theoretically possible, in practical terms it is almost never feasible. Thus, radioligand binding to a receptor is quantified in the presence of various concentrations of the unlabelled compound of interest. The concentration of the radioligand in a competition study should be about 2-3 its Kd value as determined in saturation binding; this will allow a sufficient occupancy of the receptor to obtain a strong signal and at the same time avoid that competition becomes too difficult due to high radioligand concentration. The incubation time and temperature are chosen to allow formation of equilibrium between association and dissociation with the receptor for both radioligand and competitor. Of note, a simple competition experiments does not necessarily prove a competitive nature of the interaction between unlabelled drug and receptor. If the specific radioactivity is low (tritiated) relative to the affinity of the radioligand (< 1 nM), a high assay volume (≥ 1 ml) is required to avoid ligand depletion; this is of particular importance if a receptor source with high expression density is used (e.g. expressed recombinant receptors). The number of required competitor concentrations depends on the goal of the experiment. If only a rough estimate of antagonist potency is required, 1-2 concentration per log increment will be sufficient. However, if it is the aim to test for possible subtypes or states of the receptor, 3-5 concentrations per log increment are needed. If possible, the lowest competitor concentrations in the assays should not cause any detectable inhibition, whereas the highest concentrations should completely abolish specific binding. Each experiment can be divided into different steps such as assay preparation, membrane preparation, incubation, filtration, counting of the samples and data analysis. To minimize experimental error all assays are performed at least in duplicate. Additionally, duplicates of total binding and non-specific binding should be included in the assay; the agent used for the definition of non-specific binding (NSB) should be chemically (different family) and physically (avoid combination of two lipophilic compounds) distinct from the radioligand to avoid artifacts. For discussion of specific benefits of chosen assay conditions see van Wieringen et al., (2013) (copy can be obtained from the author).
Keywords: DopamineMaterials and Reagents
- Radioligand [3H]-spiperone (e.g. PerkinElmer, catalog number: NET565250UC )
- Butaclamol (e.g. Sigma-Aldrich, catalog number: 55528-07-9 )
- Receptor-containing membrane suspension
- Competitor compounds (dopamine HCl used as example here, e.g. Sigma-Aldrich, catalog number: 62-31-7 )
- Whatman GF/C filters (e.g. PerkinElmer, catalog number: 6005174 )
- Poly(ethyleneimine) solution (PEI) (e.g. Sigma-Aldrich, catalog number: 9002-98-6 )
- Scintillation cocktail (e.g. PerkinElmer, catalog number: 6013641 )
- TRIS base
- CaCl2
- MgCl2
- Distilled water
- Ascorbic Acid
- Assay buffer (see Recipes)
- Wash buffer (see Recipes)
- 0.1% PEI (only for D3 receptor assay) (see Recipes)
- NSB solution (see Recipes)
- Dilutions of competitors compound (see Recipes)
- Radioligand solution (see Recipes)
Equipment
- 96 well plates (polysterene)
- Cell harvester (e.g. PerkinElmer)
- Ultra-Turrax (IKA, model: 0001602800 ) or similar disperser
- Water bath
- Scintillation counter
Software
- Prism (Graphpad Software) or similar
Procedure
- Before the assay
- Prepare assay and wash buffer.
- For a D3 assay: Prepare 0.1% PEI solution and pipet 100 μl/filter on the filterplate, subsequently place in refrigerator (4 °C) for at least 2 hours.
- The experiment is performed in 96 well plates (polysterene) with a total assay volume of 1,000 μl (450 μl assay buffer + 200 μl radioligand + 200 μl of assay buffer or 5 μM (+)-butaclamol + 150 μl membrane preparation).
- Prepare NSB solution.
- Prepare dilutions of competitor compound (dopamine used as example here).
- Prepare radioligand solution.
- Membrane preparation.
- Prepare receptor-containing membrane suspension according to preparation protocol. Re-homogenize suspension in small volume (< 2 ml) using short burst of Ultra-Turrax. Dilute to the desired protein concentration and to yield a total volume of about 8 ml per 48 data point experiment. The protein concentration of the membrane suspension should be chosen so that a robust specific binding signal is obtained but at the same time total binding should be < 10% (even better < 5%) of free radioligand concentration. Protein content can be assayed by a variety of essays, e.g. Bradford (1976). Prepare the membrane suspension initially in ice.
- Pre warm all solutions for 15 min in 25 °C waterbath.
- Final preparation (Table 1), add components to wells in following order:
- 250 μl assay buffer.
- 150 μl membrane suspension.
- 200 μl competitor, assay buffer (TB) or butaclamol (NSB). Use increasing concentrations of competitor.
- 200 μl assay buffer.
- Start reaction by adding 200 μl radioligand solution.
Table 1. Pipetting scheme for competitor concentrations on microtiter plateTB
TB
NSB
NSB
10
10
2 x 10
2 x 10
3 x 10
3 x 10
5 x 10
5 x 10
9
9
2 x 9
2 x 9
3 x 9
3 x 9
5 x 9
5 x 9
8
8
2 x 8
2 x 8
3 x 8
3 x 8
5 x 8
5 x 8
7
7
2 x 7
2 x 7
3 x 7
3 x 7
5 x 7
5 x 7
6
6
2 x 6
2 x 6
3 x 6
3 x 6
5 x 6
5 x 6
5
5
NSB
NSB
Total
Total
- 250 μl assay buffer.
- Prepare assay and wash buffer.
- During the assay
- Incubate for 120 min at 25 °C in water bath.
- Terminate reaction by rapid vacuum filtration over Whatman GF/C filters using a cell harvester. Wash filter 10 times with ice-cold wash buffer.
- Incubate for 120 min at 25 °C in water bath.
- After the assay
- Add aliquots of 50 μl radioligand to wells of counting plate to determine total radioactivity.
- Dry, e.g. in oven for 2 h.
- Place sticker on bottom and add scintillation cocktail (20 μl) to filters, place sticker on top of plate and count in a scintillation counter, allow adequate time (15 min) before counting samples.
- Add aliquots of 50 μl radioligand to wells of counting plate to determine total radioactivity.
- Data analysis
The data being obtained can be analyzed to yield several types of information. The following should be considered.
- Carefully inspect raw data for consistency of replicates and resist the temptation to beautify the data by eliminating apparent ‘outliers’. It is our recommendation that one should very conservative in this regard; the result of a well-designed experiment will not be heavily affected by a single outlier. In other words, if the outcome of the experiment hinges on the question whether a single data point is an outlier, the overall experiment may have been designed and/or executed poorly and should probably be repeated.
- Plot amount of binding in the absence and presence of competitor on the y-axis vs. concentration of the competitor on the x-axis. In such plots the amount of binding in the absence of competitor (total binding, TB) can be entered at a virtual concentration lower than any tested competitor concentration, and non-specific binding (NSB) at a virtual concentration higher than the highest tested competitor concentration. A representative experiment with D2 receptors may look like this.
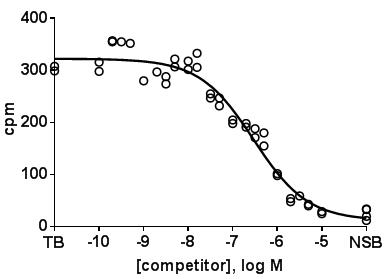
- Using all replicates without averaging them is preferable for the data analysis.
- In a well performed experiment, the concentration range of the competitor has been chosen in a way that the lowest competitor concentration does not cause any measurable inhibition and that the highest competitor concentration yields a degree of inhibition close to non-specific binding. If one of these two conditions is not met, data analysis becomes tricky as curve fitting may yield spurious values.
- Apply one of many available data analysis software packages, e.g. Prism, to fit a sigmoidal curve to the data. In most of such computer programs, you have a choice of settings which have important implications for the interpretation of the resulting parameter estimates.
- The preferred option is to let the top and the bottom of the curve to be found be the software, and the resulting values should be checked whether they are close to the experimentally determined total and non-specific binding.
- The turning point of the sigmoidal curve should be found by the software in most cases as this will yield the IC50 of the curve.
- The slope of the curve can be found by the software. Values close to unity indicate interaction with a single site. If the slope is smaller than unity, i.e. the curve is shallow, this may indicate interaction with multiple sites (see below) but does not necessarily prove it.
- If the assumption is made that an interaction with multiple sites, e.g. receptor subtypes, is possible, it can be defined that the curves should follow a monophasic or a biphasic function with each component having a slope of unity. On theoretical grounds a biphasic fit always yields a smaller residual error, as there are more fitting parameters. Therefore, a biphasic fit should only be accepted if it yields a significantly smaller residual error as judged by an F-test or similar (already implemented in most software packages for such use). If a biphasic fits is superior, it will yield IC50 values for both components and a percentage of each of the two components. e.g. for the representative experiment shown above, the sum of squares of distances of data points to the fitted curve was 24177 and 18221 for the one- and two-site fit, respectively, yielding an F value of 6.734, which indicates with a p-value of 0.0040 that the null hypothesis (single site is preferred) should be rejected and a two-site model should be preferred.
- The preferred option is to let the top and the bottom of the curve to be found be the software, and the resulting values should be checked whether they are close to the experimentally determined total and non-specific binding.
- The ability to pick up biphasic curves depends on the selectivity of the competitor for the two subtypes being present (or the intrinsic activity of the agonist, if agonist high- and low-affinity states are being analyzed) as well as the number of inhibitor concentrations being tested per log unit increment of concentration.
- The results of the curve fitting should always be inspected visually to check whether they make sense. e.g. in some cases the computer program yields estimates which are outside the range of tested competitor concentrations. Such extrapolations are highly unreliable and should not be used; rather the experiment should be repeated with a better choice of competitor concentrations.
- Estimates of IC50 depend on the concentration of radioligand in the assay, expressed as fold of its Kd value. To obtain the more informative Ki value, transformation of IC50 is necessary by the Cheng & Prusoff equation:
Ki = IC50/((L/Kd) + 1)
in which L and Kd are the concentration and affinity of the radioligand.
Notes:
- A competition binding experiment by virtue of its design cannot prove a competitive interaction between inhibitor and radioligand. This requires e.g. saturation experiments in the absence and presence of one or more inhibitor concentrations.
- As IC50 values are obtained from a log scale, the replicates from multiple experiments typically do not exhibit a Gaussian distribution on a linear scale. Hence, the average from multiple experiments should be presented as means (with SD) of –log IC50 (or –log Ki). Alternatively, the median IC50 or Ki can be presented with (asymetric) confidence intervals. Means (with SD) of linear IC50 or Ki values are inappropriate.
- A competition binding experiment by virtue of its design cannot prove a competitive interaction between inhibitor and radioligand. This requires e.g. saturation experiments in the absence and presence of one or more inhibitor concentrations.
- Carefully inspect raw data for consistency of replicates and resist the temptation to beautify the data by eliminating apparent ‘outliers’. It is our recommendation that one should very conservative in this regard; the result of a well-designed experiment will not be heavily affected by a single outlier. In other words, if the outcome of the experiment hinges on the question whether a single data point is an outlier, the overall experiment may have been designed and/or executed poorly and should probably be repeated.
Recipes
- Assay buffer
50 mM TRIS: TRIS-HCl: 6.6 g and TRIS base 970 mg/L (or only 6.04 g TRIS base/L)
5 mM KCl: 373 mg/L
2 mM CaCl2: 220 mg/L
2 mM MgCl2.6H2O: 410 mg/L
pH 7.4
Every test day prepare a fresh buffer containing 0.05% (25 mg/50 ml) ascorbic acid
Note: This buffer is optimized for detection of agonist high-affinity states of a receptor (agonist competition curves); hence, inclusion of Na+ should be avoided. If that is not within the scope of the project, a buffer without Mg2+, Ca2+ or K+ can be used.
- Wash buffer
50 mM TRIS: TRIS-HCl (33 g/5 L) and TRIS base (4.85 g/5 L)
pH 7.4
- 0.1% PEI (only for D3 receptor assay)
Prepare 0.1% solution, PEI is delivered as a 50% solution, pipet 1 ml from this with a syringe and add to 9 ml aqua dest. to get 5%, pipet 400 μl PEI 5% + 19.6 ml destilled water to get PEI 0.1%
- NSB solution
3.98 mg butaclamol HCl/10 ml assay buffer yields 1.10-3 M (prepare aliquots of this)
Dilute this 1:200 to obtain 5 μM in solution, i.e. 1 μM final concentration in asay
- Dilutions of competitors compound (dopamine used as example here)
- Molecular weight of dopamine HCl is 189.6 g/mol. Weigh 3-5 mg, calculate needed volume of assay buffer to reach target concentration; trying to start with a fixed volume and to reach a specific amount of compound is technically more difficult. In this example 3.8 mg has to be dissolved in 20 ml assay buffer to reach 10-3 M. Dilute this 2x with assay buffer to reach 5x the intended highest concentration in the assay, e.g. 5 x 10-4 M for 10-4 M in the assay.
- In the first row of below scheme place 1,800 μl assay buffer per tube, in the 2nd row (2x) 800 μl, in the 3rd row (3x) 700 μl and in the 4th row (5x) 500 μl. Amounts to be prepared may need to be increased if more than one competition experiment is to be performed.
- Dilute competitor stock solution in 1:10 steps first. Pipet from 10-4 200 μl to 5 and from 5,200 μl to 6 etc. Then, make 2x, 3x and 5x daughter solutions, pipet 200 μl in 2x, 300 μl in 3x and 500 μl in 5x. Do this also for 8, 7, 6, 5 and 4. (Table 2)
Table 2. Dilution scheme for competitor working solutions
5x
5x
5x
5x
5x
NSB 3x
3x
3x
3x
3x
2x
2x
2x
2x
2x
9
8
7
6
5
4
- Molecular weight of dopamine HCl is 189.6 g/mol. Weigh 3-5 mg, calculate needed volume of assay buffer to reach target concentration; trying to start with a fixed volume and to reach a specific amount of compound is technically more difficult. In this example 3.8 mg has to be dissolved in 20 ml assay buffer to reach 10-3 M. Dilute this 2x with assay buffer to reach 5x the intended highest concentration in the assay, e.g. 5 x 10-4 M for 10-4 M in the assay.
- Radioligand solution
The intended radioligand concentration in the assay should be about 2-3x Kd (Kd spiperone in transfected cells according to literature ≈ 0.05 nM, 0.35 nM and 0.07 nM for D2, D3 and D4, respectively). The stock solution to be prepared needs to be 5x the assay concentration. Thus, the stock solution should be 10-15x Kd. The following is an example calculation based on a specific activity of 16.2 Ci/mmol radioactive of the radioactive stock solution for a D2 receptor assay. This needs to be adapted for the other subtypes based on their Kd values and for each batch of radioligand and its specific activity. Prepare about 10 ml of stock solution per planned 48 data point experiment. Stock solution can be prepared in 50 ml tube with screw cap.
[3H]-spiperone concentration = 1 mCi/ml/(16.2 Ci/mmol x 1,000 mCi/Ci) = 61.7 μM
Highest concentration needed (3 x Kd x 5) = 3 x 0.05 x 5 = 0.75 nM
10 ml x 0.75 nM = 0.0075 nmol [3H]-spiperone needed
(0.0075 nmol)/61.7 x 103 nM = 0.12 μl of [3H]-spiperone solution needed for 10 ml
Thus 1 μl [3H]-spiperone solution in 83.3 ml assay buffer to give a concentration of 0.75 nM
A free tool for such calculations can be found at www.graphpad.com/quickcalcs/chemMenu/.
Acknowledgments
This protocol is the adaptation of a protocol originally published by Levant (2007).
References
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
- Levant, B. (2007). Characterization of Dopamine Receptors. Curr Protoc Pharmacol 36:1.6.1–1.6.15.
- van Wieringen, J. P., Booij, J., Shalgunov, V., Elsinga, P. and Michel, M. C. (2013). Agonist high- and low-affinity states of dopamine D(2) receptors: methods of detection and clinical implications. Naunyn Schmiedebergs Arch Pharmacol 386(2): 135-154.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wieringen, J. V. and Michel, M. C. (2013). [3H]-Spiperone Competition Binding to Dopamine D2, D3 and D4 Receptors. Bio-protocol 3(20): e944. DOI: 10.21769/BioProtoc.944.
Category
Neuroscience > Sensory and motor systems
Biochemistry > Protein > Interaction > Protein-ligand interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.