- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Ultra-low Background DNA Cloning System

Published: Vol 3, Iss 16, Aug 20, 2013 DOI: 10.21769/BioProtoc.874 Views: 18668
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
We have developed a method to clone DNA fragments into the E. coli plasmid vectors with almost 100% efficiency (Goto and Nagano, 2013). This method is based on highly efficient yeast-based in vivo cloning, and the subsequent cloning of the constructed plasmids into E. coli. Our method is useful for various applications: multifragment DNA cloning, cloning of large DNA fragments, and cloning into large plasmid vectors. Furthermore, the sites at which DNA fragments are joined are not always located at the restriction ends in the plasmid vector, thus making the cloning method more flexible. Our system does not require manipulation for assembling or joining DNA fragments in a test tube, the efficiency of which may sometimes depend on the reaction conditions or the skills of the person performing the procedure. Therefore, both success rate and efficiency are extremely high. However, our system has a disadvantage in that it requires 2 steps for transformation. Our method is an improved version of previously developed methods (Iizasa and Nagano, 2006; Nagano et al., 2007). Next figure shows the flowchart of our method.
Keywords: Plasmid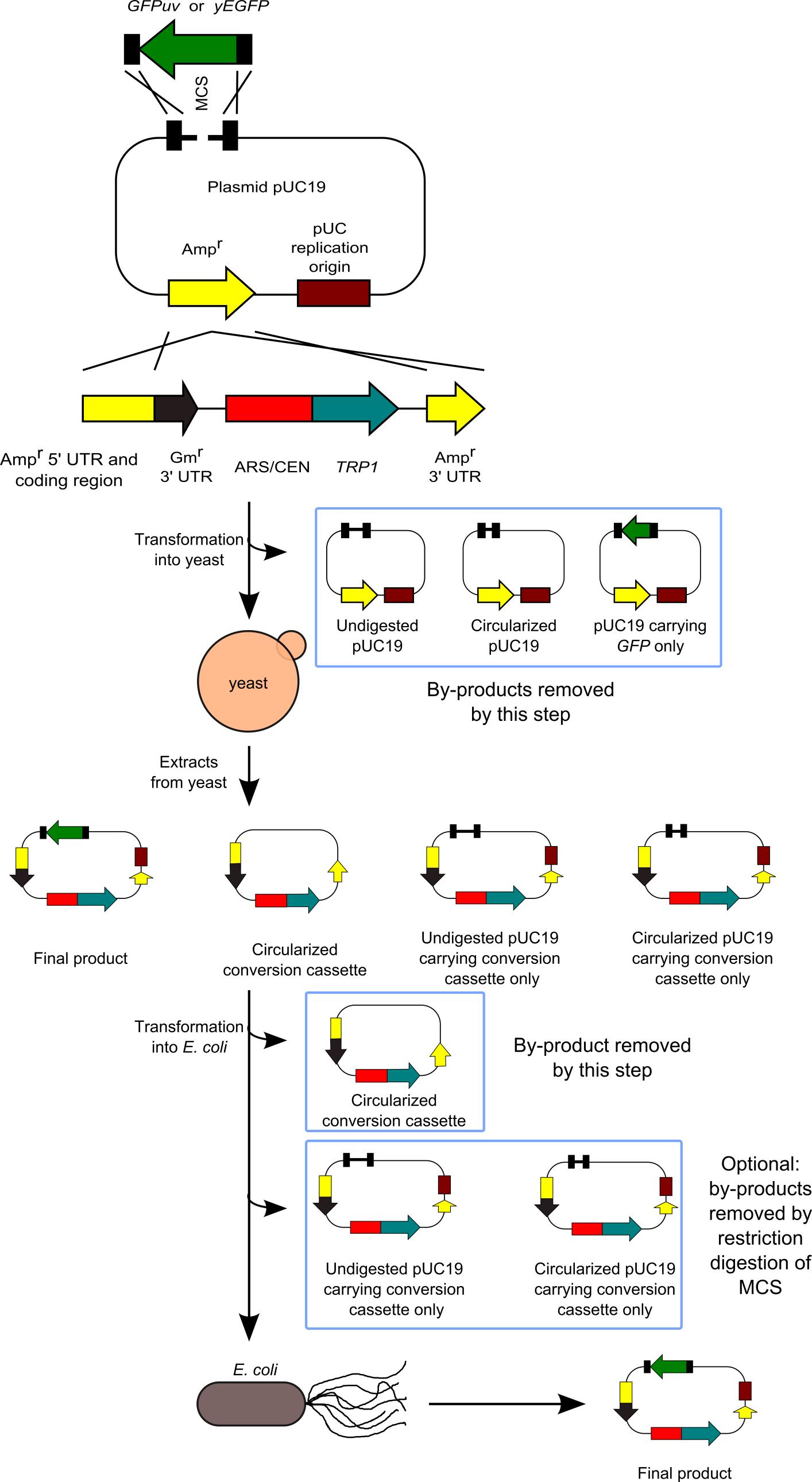
Figure 1. Flowchart of our method
Materials and Reagents
- Plasmids/nucleic acids
- Plasmid pSU32 (http://www.iac.saga-u.ac.jp/lifescience/su32/index.htm) (Figure 2)
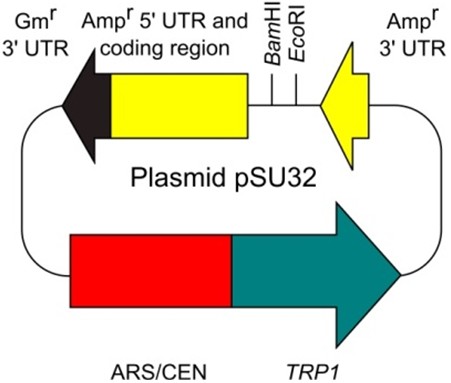
Figure 2. Plasmid pSU32 - E. coli plasmid vector containing the Ampr gene (bla gene) (e.g., pUC, pBluescript, and the other many types of commonly-used plasmid vectors)
- DNA fragment to be cloned
- PCR primers (the pSU30-14 and pSU30-23 pair for the preparation of the conversion cassette SU32, and primer pair for the preparation of the DNA fragment to be cloned)
the primer pair for the preparation of the conversion cassette SU32 pSU30-14 5'-CAGGTGGCACTTTTCGGGGAAATGTG-3' pSU30-23 5'-ATCAAAAAGGATCTTCACCTAGATCCTTTTAAATTAAAAATGA AGTTTTAAATCAATCTAAAGTATATATGAGTAAACT-3' Example: Primer pair for the preparation of the DNA fragment to be cloned
(in this case, the GFPuv gene was cloned into pUC19 plasmid. The crossover regions (the sequences to be joined) are underlined).pUCGFPF 5'-GAGCGGATAACAATTTCACACAGGAAACAGCTATGGCTAGCA AAGGAGAAGAACT-3' pUCGFPR 5'-TTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGTTATTTG TAGAGCTCATCCA-3'
- Plasmid pSU32 (http://www.iac.saga-u.ac.jp/lifescience/su32/index.htm) (Figure 2)
- Kits
- (Optional) PCR Purification kit
In our paper (Goto and Nagano, 2013), we used the MonoFas DNA Purification Kit I (GL Sciences, Tokyo, Japan). However, this kit currently does not work well in our laboratory. Instead, we are now using the NucleoSpin Gel and PCR Clean-up kit (MACHEREY-NAGEL, Düren, Germany). - Plasmid DNA purification kit for E. coli
We used a Mini-M Plasmid DNA Extraction System (Viogene, Taipei, Taiwan) (Goto and Nagano, 2013). - Suspension buffer (in the case of Mini-M Kit, we use 200 μl of MX1 buffer)
- (Optional) PCR Purification kit
- Enzymes
- Proofreading DNA polymerase such as Prime STAR GXL DNA Polymerase (TAKARA BIO, Ohtsu, Japan)
- Restriction enzymes (New England Biolabs)
- Proofreading DNA polymerase such as Prime STAR GXL DNA Polymerase (TAKARA BIO, Ohtsu, Japan)
- Cells
- Yeast cells (commonly used strains containing the genotype trp1, such as YPH499)
- Electro-competent E. coli cells
We used the E. coli HST08 Premium Electro-Cells (TAKARA BIO, Ohtsu, Japan) (Goto and Nagano, 2013). However, these cells currently do not work well in our laboratory. Instead, we are now making electrocompetent cells ourselves according to Molecular Cloning (4th edition, pages 177-182).
- Yeast cells (commonly used strains containing the genotype trp1, such as YPH499)
- Plates
- LB agar containing 100 μg/ml ampicillin (Molecular Cloning, 4th edition, page 1100)
- Synthetic complete plates lacking tryptophan (The Gietz Lab, University of Manitoba, http://home.cc.umanitoba.ca/~gietz/ or Gietz and Woods, 2002)
- Ampicillin (sodium salt)
- LB agar containing 100 μg/ml ampicillin (Molecular Cloning, 4th edition, page 1100)
- Others
- Plating beads
- Acid-washed glass beads (350-500 μm)
We are making acid-washed glass beads ourselves. However, Acid-washed glass beads (425–600 μm) can be obtained from SIGAMA-ALDRICH (catalog number: G8772). - Agarose gel (Molecular Cloning, 4th edition, pages 94-98)
- Materials and Reagents required for yeast transformation (The Gietz Lab, University of Manitoba, http://home.cc.umanitoba.ca/~gietz/ or Gietz and Woods, 2002)
- Plating beads
Equipment
- Thermal Cycler
- Agorose gel electrophoresis device
- Centrifuge
- Optional: Picofuge
- Incubators (30 °C and 37 °C)
- Vortex machine
- Optional: FastPrep FP100A (MP Biomedical)
- Heat block (70 °C)
- Electroporation device and cuvette
- Polyacrylamide gel electrophoresis devices
- Glass beads (350-500 μm)
Procedure
- Preparation of conversion cassette SU32
- Prepare the conversion cassette SU32 by PCR using a plasmid pSU32 as template. To obtain this template, visit the following webpage.
http://www.iac.saga-u.ac.jp/lifescience/su32/index.htm - For PCR amplification, the primer pair was pSU30-14 and pSU30-23 (40 cycles of 98 °C for 10 sec, 55 °C for 15 sec, and 68 °C for 75 sec using Prime STAR GXL DNA Polymerase) (Goto and Nagano, 2013). When we conduct the PCR reaction, we use Prime STAR GXL DNA Polymerase, but you can use the other proofreading DNA polymerases.
Note: Although we purified the amplified conversion cassette in our paper (Goto and Nagano, 2013), this purification step is not essential (purification is required for DNA quantification). - Confirm the amplification by 1% agarose gel electrophoresis. The amplicon size is about 2.7 kb (2,651 bp) (Figure 3).
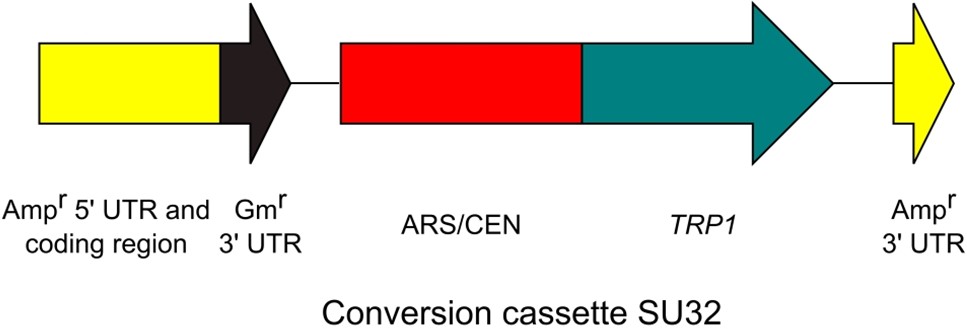
Figure 3. Conversion cassette SU32
- Prepare the conversion cassette SU32 by PCR using a plasmid pSU32 as template. To obtain this template, visit the following webpage.
- Preparation of the linearized E. coli plasmid vector
- Digest the E. coli plasmid vector within the crossover region by using restriction enzyme.
Notes:- The required restriction ends can be located anywhere within the crossover regions of the vector. You can use both blunt-end restriction enzyme and sticky-end restriction enzyme, because restriction ends are not positions where joining reactions occur.
- This E. coli plasmid vector must contain the Ampr gene.
- PCR amplification of the E. coli plasmid vector is another way to prepare the linearized E. coli plasmid vector.
- Although we purified the digested plasmid in our paper (Goto and Nagano, 2013), this purification step is not essential.
- The required restriction ends can be located anywhere within the crossover regions of the vector. You can use both blunt-end restriction enzyme and sticky-end restriction enzyme, because restriction ends are not positions where joining reactions occur.
- Confirm the digestion by 1% agarose gel electrophoresis.
- Next Figure shows one of the examples of the restriction digestion (Figure 4 also shows how to join the DNA fragments).
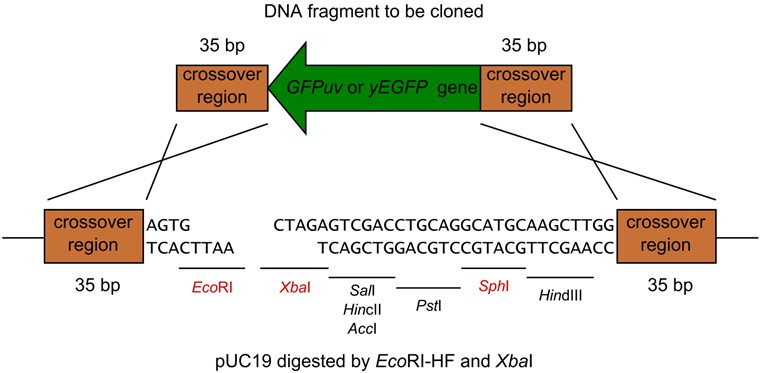
Figure 4. pUC19 digested by EcoRI-HF and XbaI
- Digest the E. coli plasmid vector within the crossover region by using restriction enzyme.
- Preparation of the DNA fragment to be cloned.
- Amplify a DNA fragment of interest by PCR using proofreading DNA polymerase such as Prime STAR GXL DNA Polymerase.
Notes:- PCR primers should be designed to carry the sequences with more than 20 bp of homology to the crossover region. We often use the sequences with 30-40 bp of homology to the crossover region. For the highest efficiency, we recommend you to use PCR primers purified by polyacrylamide gel electrophoresis. However, the PAGE purification is not essential.
- Although we purified the DNA fragment of interest in our paper (Goto and Nagano, 2013), this purification step is not essential.
- PCR primers should be designed to carry the sequences with more than 20 bp of homology to the crossover region. We often use the sequences with 30-40 bp of homology to the crossover region. For the highest efficiency, we recommend you to use PCR primers purified by polyacrylamide gel electrophoresis. However, the PAGE purification is not essential.
- Confirm the amplification by agarose gel electrophoresis.
- Amplify a DNA fragment of interest by PCR using proofreading DNA polymerase such as Prime STAR GXL DNA Polymerase.
- Transformation of DNA fragments into yeast.
- Transform these 3 DNA fragments into yeast by TRAFO protocol (Gietz and Woods, 2002). TRAFO protocol is available on the following webpage.
http://home.cc.umanitoba.ca/~gietz/
Notes:- In our laboratory, we perform the transformation procedure at half or quarter scale of this method.
- The molar ratio of the 3 DNA fragments was 1:1:1. However, this ratio is not important (Goto and Nagano, 2013). (Ten-fold variation of the ratio probably does not affect the efficiency.) Required amount of the conversion cassette SU32 is < 50 ng.
- In our laboratory, we perform the transformation procedure at half or quarter scale of this method.
- Select the transformants by incubating yeast cells on synthetic complete plates lacking tryptophan at 30 °C for 2 or 3 days.
- Transform these 3 DNA fragments into yeast by TRAFO protocol (Gietz and Woods, 2002). TRAFO protocol is available on the following webpage.
- Purification of plasmid from yeast.
Note: To purify plasmid DNAs from yeast, we use a Mini-M Plasmid DNA Extraction System, but you can use the other Plasmid DNA Extraction Kit for E. coli.- Scrape all colonies from the plates with plating beads, and collect yeast cells by the centrifuge. Using the microfuge, centrifuge cells at 14,000 x g for 5 sec (We often use the picofuge at a maximum speed for 30 sec). Remove the supernatant.
- Add the suspension buffer (in the case of Mini-M Kit, we use 200 μl of MX1 buffer), and suspend the yeast cells by vortexing.
- Scoop equal amounts of acid-washed glass beads (350–500 μm) to the suspended solution using 1.5 ml tube, add glass beads into tube containing the suspended solution, and then disrupt yeast cells by shaking 3 times for 20 sec at speed 5.5 using the FastPrep FP100A. Alternatively, vortex 5 times for 1 min at maximum speed. Next pictures show these experimental steps.

- Add denaturation buffer (in the case of Mini-M Kit, we use 250 μl of MX2 buffer), and gently mix.
- Punch a hole in the bottom of the tube by needle (18 gauge). Next pictures show these experimental steps.


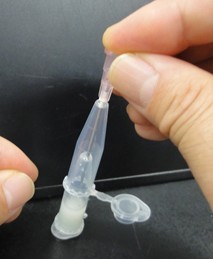
- Attach another tube to the bottom of the tube, and centrifuge them to recover the denatured solution in the bottom tube. Next pictures show these experimental steps.
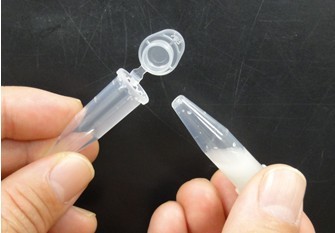


- For subsequent procedures, follow the manufacturer’s instructions. However, use elution solution preheated to 70 °C. Because DNA concentration of the eluted solution is low, you cannot determine the concentration of DNA by agarose gel electrophoresis or spectrophotometer. However, the DNA concentration of the eluted solution is sufficient for next procedures. Therefore, the condensation of the eluted solution is not required.
- Scrape all colonies from the plates with plating beads, and collect yeast cells by the centrifuge. Using the microfuge, centrifuge cells at 14,000 x g for 5 sec (We often use the picofuge at a maximum speed for 30 sec). Remove the supernatant.
- Pretreatment of the eluted solution (Optional)
- For the highest efficiency, digest 5 μl of the recovered plasmids (the eluted solution) with small amounts (e.g. 0.2 μl) of an appropriate restriction enzyme by directly adding the enzyme to the eluted DNA solution.
Note: Because the principle of this method is complicated, read our paper (Goto and Nagano, 2013) carefully. Do not add an enzyme reaction buffer, because the salt may affect the next electroporation step. - Incubate the tube at an appropriate temperature for more than 1 h.
- For the highest efficiency, digest 5 μl of the recovered plasmids (the eluted solution) with small amounts (e.g. 0.2 μl) of an appropriate restriction enzyme by directly adding the enzyme to the eluted DNA solution.
- Transformation of the eluted solution into E. coli.
Transform the eluted solution into E. coli electro-competent cells by electroporation and plate them on LB agar containing 100 μg/ml ampicillin. - Analyze E. coli transformants by various methods.
Acknowledgments
This protocol was adapted from previously published paper, Goto and Nagano, (2013). Part of this work was supported by A-STEP (FS-stage) from the Japan Science and Technology Agency (JST) and by a Grant-in-Aid for Challenging Exploratory Research from the Japan Society for the Promotion of Science.
References
- Gietz, R. D. and Woods, R. A. (2002). Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350: 87-96.
- Goto, K. and Nagano, Y. (2013). Ultra-low background DNA cloning system. PLoS One 8(2): e56530.
- Iizasa, E. and Nagano, Y. (2006). Highly efficient yeast-based in vivo DNA cloning of multiple DNA fragments and the simultaneous construction of yeast/Escherichia coli shuttle vectors. Biotechniques 40(1): 79-83.
- Nagano, Y., Takao, S., Kudo, T., Iizasa, E. and Anai, T. (2007). Yeast-based recombineering of DNA fragments into plant transformation vectors by one-step transformation. Plant Cell Rep 26(12): 2111-2117.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nagano, Y. and Goto, K. (2013). Ultra-low Background DNA Cloning System. Bio-protocol 3(16): e874. DOI: 10.21769/BioProtoc.874.
Category
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.