- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation, Purification, and Culture of Primary Murine Microglia Cells

Published: Vol 3, Iss 1, Jan 5, 2013 DOI: 10.21769/BioProtoc.314 Views: 30880
Original research article
The authors used this protocol in:

Jul 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The following is a detailed protocol for the isolation, purification and culture of murine brain microglia cells using neutral enzyme digestion and shaking. The protocol below is designed to isolate and culture a large number of purified inactivated microglia cells. Neutral enzyme digestion allows for minimal cellular damage and a higher cell recovery compared to other mechanical dissociative methods such as mincing. Cells cultured using this method will display the size and morphological features of microglia cells.
Keywords: IsolationMaterials and Reagents
- Five to eight fetal (1-3 days old) BALB/C mice (Harlan)
- Ice
- DMEM(Sigma-Aldrich, catalog number: D5796 )
- Fetal bovine serum heat inactivated (FBS) (Sigma-Aldrich, catalog number: F9665 )
- D-Hank’s balanced salt solution without phenol red (Life Technologies, InvitrogenTM, catalog number: H6648 )
- Trypsin / EDTA (0.05%/0.02%) (Life Technologies, InvitrogenTM, catalog number: 25300054 )
- Alcohol (70%)
- Poly-D-lysine solution (10 μg/ml) (Sigma-Aldrich, catalog number: L202 )
- 70% Alcohol
- Physiological saline (0.9%) (Sigma-Aldrich, catalog number: 07982-100TAB-F )
- Trypsin (0.25%) (Life Technologies, InvitrogenTM, catalog number: 25200-072 )
Two different concentrations of DMEM with FBS are used in order to maximize growth. The logic behind this is that the micrglia are more ‘sensitive’ after enzymic digestion. Therefore the use of DMEM with a higher concentration of FBS (i.e. 15%) provides additional growth factors and buffering agents after disaggregation and its use prior to enzymic expose is to allow the cells to acclimatize to this higher concentration and to optimize the cells under this improved growing condition. DMEM +FBS 10% is sufficient for culturing at all other periods.
Equipment
- Straight scissors
- Curved scissors
- Corneal scissors
- Straight forceps (small, x2)
- Curved artery forceps (x3)
- Beaker (100 ml, x2)
- Culture dish (30 mm, x2)
- Culture flask (25 cm2 and 75 cm2)
- Centrifuge Falcon tubes (15 ml and 50 ml)
- Serological pipettes (2 ml and 5 ml)
- Plastic transfer pipettes (3.5 ml)
- 70-μm nylon cell strainer
- Sterile gauze
- Incubator (37 °C; 5% CO2; humidified)
- Refrigerated centrifuge
- Magnifying glass and microscope
- UV light
- Culture flask with mixed glia cells
- Parafilm
- Alcohol burner
- Orbital shaker/Belly dancer (37 °C)
- 24-well tissue culture plate pre-coated with poly-D-lysine
Procedure
Caution: Bio-safety practices (Level II) should be adhered to during handling of both animals and cell cultures.
Note: All procedures are performed under aseptic conditions.
- Stage 1: Isolation and culture of murine microglia cells via enzymatic digestion.
Enzymatic disaggregation of mouse brain tissue with trypsin provides a high yield of microglia cells without any detectable cell activations.- The base of a 75 cm2 culture flask is coated with 5 ml poly-D-lysine (10 μg/ml) and allowed to set overnight at room temperature (note: Poly-D-lysine assists in the cells adhering to the flask).
- On the day of the experiment all excess poly-D-lysine is removed as any excess solution is caustic to the microglia cells. The flask is then washed once with PBS. In addition the cleaned culture cabinet/hood is exposed to an additional 30 min of UV light to maximize the sterilization process.
- Five to eight fetal BALB/c mice (1-3 days old) are culled by cutting both carotid arteries (without decapitating the mouse; otherwise subsequent handling is made difficult) with a pair of scissors and the heads are placed in a beaker with 70% alcohol for 15-30 sec. Step (3) to (g) is represented by Figure 1 (see below).
- The decapitated heads are removed and all excess blood and alcohol is removed using a piece of sterile gauze.
- Next the skin over the skull is cleared under aseptic technique. This is achieved by making a crucifix incision over the cranium (transverse incision at the base of the skull and the vertical incision along the midline of the skull up to the anterior part of the skull). A new straight forceps is then used to peel away the skin on the scalp to fully expose the skull.
- The lambda and sagittal sutures are identified. A pair of corneal scissors are used to cut along the suture lines. A 3rd pair of straight forceps are then used to ‘peel’ the skull away (note: The skull of a mouse this age is very soft and pliable) and to fully expose the brain (note: The following 2 steps must be performed on ice to minimize cell loss).
- A new curved forceps is then inserted under the base of the brain (taking care not to damage the brain). Gently lift the brain away from the skull base. Place the brain onto a small culture dish that contains a small volume of D-Hanks balanced salt solution.
- The meningeal lining (dura and arachnoid layers) is then gently removed using two small straight forceps under a magnifying glass. The cleaned brain is then placed in a new culture dish with 2 ml of trypsin / EDTA (0.05%/0.02%). The brain is then minced into a fine slurry using a pair of corneal scissors.
- The brain / trypsin suspension is transferred gently into a 15 ml centrifuge tube using a 5 ml serological pipette and the same volume of trypsin / EDTA (0.05%/0.02%) is added. The tube is then maintained in the incubator at 37 °C in a humidified 5% CO2 atmosphere for 8-10 min to allow for enzymatic disaggregation.
- After the allocated time, DMEM with 10% FBS is added to the suspension (1:1 ratio) to neutralize the trypsin. The single cell suspension is gently agitated by moving the solution up and down 20 times using a 3.5 ml plastic transfer pipette with care being taken to avoid bubble formation.
- The suspension is allowed to stand for a minute at room temperature to allow any large pieces to precipitate. This supernatant is passed through a 70 μm nylon cell strainer into another 100 ml beaker. The process of agitation using a 3.5 ml plastic transfer pipette with more DMEM with 10% FBS (equal volume to the supernatant) and straining is repeated twice.
- All the filtrated fluid is then gently transferred into a 50 ml centrifuge tube using a serological pipette. This is then centrifuged in a cooled (4 °C) centrifuge at 190 x g for 8 min.
- The supernatant is then discarded and the cell pellet is resuspended in 15 ml of DMEM containing 15% FBS.
- The resuspended cells are added to the poly-D-lysine coated flask and incubated at 37 °C in a humidified 5% CO2 atmosphere. After 3 days, the DMEM is replaced with a fresh batch of 15 ml of DMEM containing 10% FBS. After this, the medium is changed every alternate day using a 1:1 mix of fresh DMEM containing 10% FBS and the supernatant of the medium of the flask in order to remove any dead cells and retain existing growth factors previously released by the cells. This is achieved by removing the existing 15 ml via a serological pipette into a 50 ml centrifuge tube period followed by an immediate replacement with 7.5 ml of fresh DMEM with 10% FBS. The ‘old’ medium is then centrifuged to remove dead cells at 190 x g. To make the 15 ml required for the flask, 7.5 ml of the centrifuged supernatant is added. This process can be repeated for up to 10-14 days.
- When the cells are approximately 80-90% confluent, they can be used for purification.
- The base of a 75 cm2 culture flask is coated with 5 ml poly-D-lysine (10 μg/ml) and allowed to set overnight at room temperature (note: Poly-D-lysine assists in the cells adhering to the flask).
- Stage 2: Purification of murine microglia cells from the mixed glia cell culture
- Once the mixed glia cell culture achieves a 90% confluence level (after 10-14 days) the microglia cells can be detached from the flask using the heated orbital shaker/belly dancer machine at 240 rpm for 1.5 - 2 h (37 °C). Twenty-hour hours prior to disaggregation, the medium is replaced with fresh DMEM containing 15% FBS.
- The next day the flask is placed on the heated orbital shaker at 200-240 rpm/min at 37 °C for 2 h.
- The suspension is transferred into a 50 ml centrifuge tube and then centrifuged at 190 x g for 8 min.
- The supernatant is discarded and the cell pellet is resuspended in fresh DMEM containing 15% FBS.
- The cells are then seeded onto a 24-well tissue culture plate that has been pre-coated with 5 ml of poly-D-lysine (10 μg/ml). The cells are distributed into 7.5 x 104 cells/well and incubated at 37 °C in a humidified 5% CO2 atmosphere. The medium is replaced every 2-3 days with fresh DMEM containing 10% FBS. Cell growth is observed regularly using a microscope (Figure 2). The cellular yield is 1 x 106 microglia / mouse.
- The original mixed glia cell flask is replenished with 10 ml fresh DMEM containing 10% FBS. After this, the medium is changed every 2-3 days using a 1:1 mix of fresh DMEM containing 10% FBS and the supernatant of the medium of the flask as described above. The purification process described above can then be repeated a week later. After plating the cells in the 24-well plate, the cells must be used within a week (optimal for the first 3 days). The cell purity can be assessed via flow cytometry using the anti-CD11b monoclonal antibody.
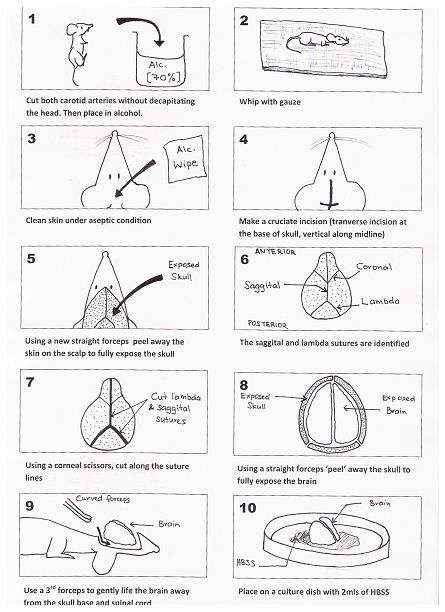
Figure 1. Method for the removal of the brain from a 1-3 day old mouse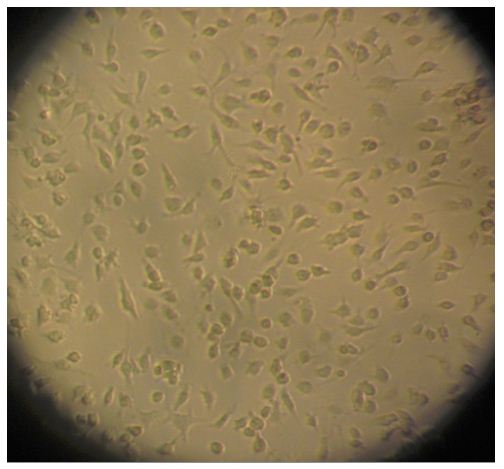
Figure 2. Microglia on light microscopy
- Once the mixed glia cell culture achieves a 90% confluence level (after 10-14 days) the microglia cells can be detached from the flask using the heated orbital shaker/belly dancer machine at 240 rpm for 1.5 - 2 h (37 °C). Twenty-hour hours prior to disaggregation, the medium is replaced with fresh DMEM containing 15% FBS.
Acknowledgments
This work was supported by National Natural Science Foundation of China (grants 30972718 and 81273242), Natural Science Foundation of Jiangsu Province (grant BK2012605), and Jiangsu Province Program of Innovative and Entrepreneurial Talents (2011-2014).
References
- Chen, X., Quinn, E. M., Ni, H., Wang, J., Blankson, S., Redmond, H. P., Wang, J. H. and Feng, X. (2012). B7-H3 participates in the development of experimental pneumococcal meningitis by augmentation of the inflammatory response via a TLR2-dependent mechanism. J Immunol 189(1): 347-355.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chen, X., Zhang, Y., Sadadcharam, G., Cui, W. and Wang, J. H. (2013). Isolation, Purification, and Culture of Primary Murine Microglia Cells. Bio-protocol 3(1): e314. DOI: 10.21769/BioProtoc.314.
Category
Immunology > Immune cell isolation > Glial cell
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.