- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Purification of Schwann Cells from Spinal Nerves of Neonatal Rat
Published: Vol 7, Iss 20, Oct 20, 2017 DOI: 10.21769/BioProtoc.2588 Views: 12967
Reviewed by: Sébastien GillotinAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Primary cultured Schwann cells (SCs) are widely used in the investigation of the biology of SC and are important seed cells for neural tissue engineering. Here, we describe a novel protocol for harvesting primary cultured SCs from neonatal Sprague-Dawley (SD) rats. In the present protocol, dissociated SCs are isolated from the spinal nerves of neonatal rats and purified by the treatment of cytosine arabinoside (AraC).
Keywords: Schwann cellBackground
SCs are the glial cells of the peripheral nervous system (PNS). Isolation and purification of primary SCs are crucial steps for studying the biology of SC. In addition, purified primary cultured SCs are important seed cells for neural tissue engineering. To date, various methods of culturing SCs have been reported based on the method of Brockes (Brockes et al., 1979). By reported methods, sciatic nerves are mostly used for SC isolation because they are large in size and can be easily obtained. However, SCs from sciatic nerves are easily contaminated with fibroblasts because the connective tissue is difficult to be cleared off. Especially the epineurium and perineurium are the main source of fibroblasts. Without special treatment, contaminating fibroblasts proliferate much faster than SCs and will soon be the predominant cells in the cultures. In the past decades, numerous purification methods have been developed for isolating SCs from the contaminating fibroblasts. The details of these purification methods included single or combination of antimitotic treatment (Wood, 1976), antibody-mediated cytolysis (Brockes et al., 1979), immunoselection (Assouline et al., 1983; Vroemen and Weidner, 2003), repeated explantation (Oda et al., 1989), cold jet technique (Haastert et al., 2007), differential adhesion (Pannunzio et al., 2005) and differential detachment (Jin et al., 2008). These purification methods involve either complicated techniques with high cost or long harvested period with low cell yield. Therefore, obtaining a large number of purified SCs is still a challenging work for basic research and further clinical use. Here, we describe a method that uses spinal nerves from neonatal SD rats as a cell source to efficiently obtain highly purified SCs in a short period.
Materials and Reagents
- Pipette tips (Corning, Axygen®, catalog numbers: T-1000-B-R-S , T-200-Y-R-S )
- Cell culture dish (35 x 10 mm) (3.5-cm dish) (Corning, catalog number: 430165 )
- 1.5 ml centrifuge tubes (Corning, Axygen®, catalog number: MCT-150-C )
- 50 ml centrifuge tubes (Corning, catalog number: 430290 )
- 100 µm cell strainer (Corning, catalog number: 431752 )
- Neonatal Sprague-Dawley (SD) Rat (Postnatal 2-4 days, P2-4)
- Distilled water
- 75% ethanol
- Hank’s balanced salts solution (HBSS) (Thermo Fisher Scientific, GibcoTM, catalog number: C14175500BT )
- Poly-L-lysine hydrobromide (PLL) (Sigma-Aldrich, catalog number: P1274 )
- 0.25% trypsin-EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 25200072 )
- Fetal bovine serum (FBS) (Mediatech, catalog number: 35-076-CV )
- Dulbecco’s modification of Eagle’s medium/Ham’s F-12 50/50 Mix (DMEM/F12) (Mediatech, catalog number: 10-092-CV )
- Cytosine Arabinoside (AraC) (Sigma-Aldrich, catalog number: C1768 )
- Forskolin (Sigma-Aldrich, catalog number: F6886 )
- Recombinant Human Heregulin β-1 (Heregulin) (PeproTech, catalog number: 100-03 )
- Dimethyl sulfoxide (DMSO) (MP Biomedicals, catalog number: 196055 )
- Penicillin/streptomycin (Pen/Strep) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Phosphate-buffered saline (PBS) (Beyotime Biotechnology, catalog number: C0221A )
- Paraformaldehyde (PFA) (Guangdong Guanghua Sci-Tech, catalog number: 1.17767.014 )
- Gelatin (Sigma-Aldrich, catalog number: G7041 )
- Triton X-100 (Sigma-Aldrich, catalog number: V900502 )
- 4,6-Diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, catalog number: D9542 )
- Anti-glial fibrillary acidic protein antibody produced in rabbit (GFAP) (Sigma-Aldrich, catalog number: G9269 )
- Anti-S-100 protein antibody, clone 15E2E2, produced in mouse (S100) (Merck, catalog number: MAB079-1 )
- Anti-nerve growth factor receptor antibody, p75, produced in rabbit (P75) (Merck, catalog number: AB1554 )
- Alexa Fluor® 488 goat anti-mouse IgG (H+L) (Thermo Fisher Scientific, Invitrogen, catalog number: A-11001 )
- Alexa Fluor® 488 goat anti-rabbit IgG (H+L) (Thermo Fisher Scientific, Invitrogen, catalog number: A-11008 )
- 100x PLL (see Recipes)
- 10% FBS (see Recipes)
- 1,000x AraC (see Recipes)
- 500x heregulin (see Recipes)
- 10,000x forskolin (see Recipes)
- SC culture medium (see Recipes)
- 4% paraformaldehyde (PFA) (see Recipes)
- 0.1% Triton X-100 (see Recipes)
- Blocking buffer (see Recipes)
- 1,000x DAPI (see Recipes)
Equipment
- Pipettes (Eppendorf, model: Research Plus® , 200 μl, 1000 μl)
- CO2 incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: Model 3100 Series , catalog number: 3111)
- Tissue culture hood (AIRTECH, catalog number: SW-CJ-1F )
- Surgical scissors and forceps (RWD Life Science, catalog numbers: S14001-15 , F13024-13 , S12003-09 , F13029-10 , see Figure 1A)
Note: S14001-15 and F13024013 are scissors and forceps used for decapitation, S12003-09 and F13029-10 are scissors and forceps used for skin dissection. - Spring scissors (66 Vision, catalog number: 54053B , see Figure 1B)
- Fine forceps (Fine Science Tools, Dumont, model: #5, catalog number: 11252-23 , with tips of 0.1 x 0.06 mm, see Figure 1B)
- Superfine forceps (Fine Science Tools, Dumont, model: #5, catalog number: 11252-20 , with tips of 0.05 x 0.02 mm, see Figure 1B)
- Dissecting board, needles and ice packs
- Water bath (Ningbo Scientz Biotechnology, model: GH-15 )
- Stereomicroscope (Olympus, model: SZ61 )
- Centrifuge (Eppendorf, model: 5430 )
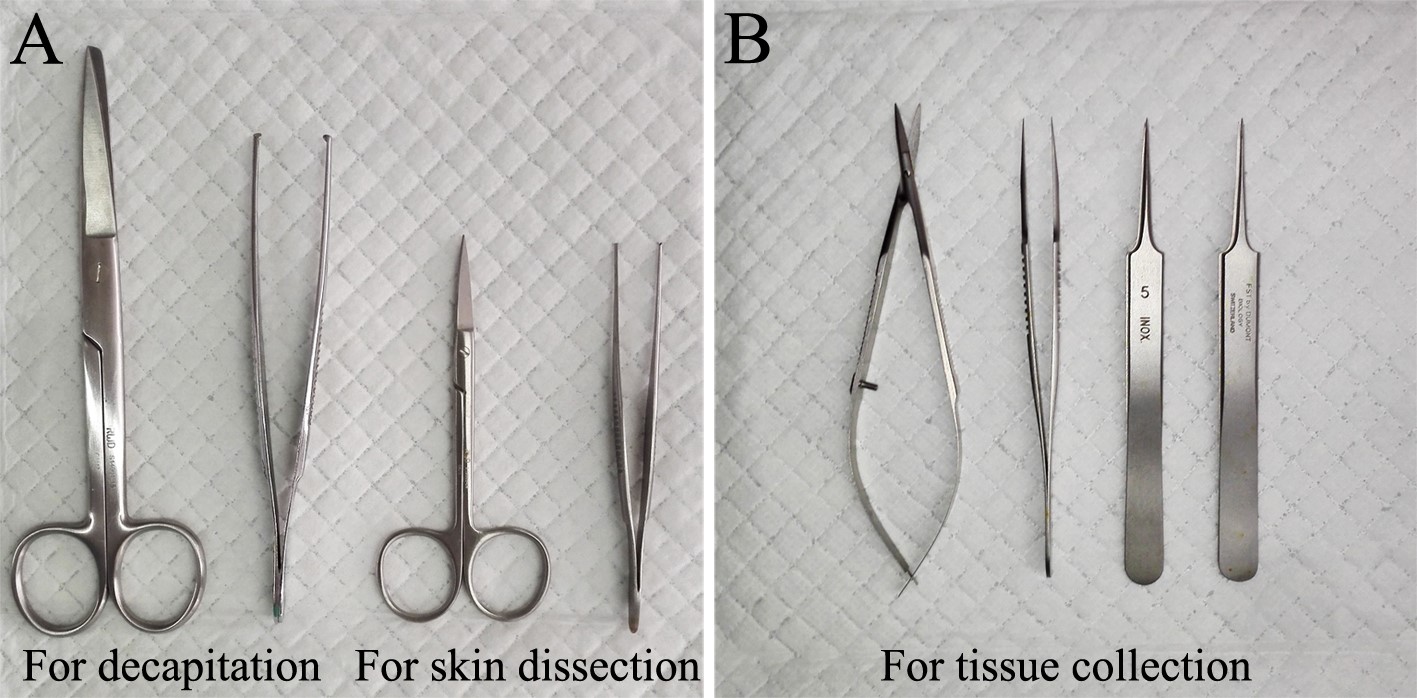
Figure 1. Dissection tools. A. Surgical scissors and forceps used to decapitate the rat and to dissect skin. B. Spring scissors and fine forceps used to separate muscle, open vertebral laminae and collect spinal nerves.
Procedure
- Preparation
- Coat a 3.5-cm dish with 1 ml PLL solution (0.1 mg/ml in distilled water, see Recipes) at 37 °C in a CO2 incubator overnight.
- Immerse the scissors and forceps in 75% ethanol for 30 min to sterilize them and then expose them to UV light for 30 min to air dry them.
- Sterilize the dissection room (in where the rat to be dissected) by UV light for 30 min.
- Remove PLL solution through aspiration and thoroughly rinse the dish surface with distilled water twice. Air dry the PLL-coated dish in the tissue culture hood before dissection.
- Prepare two 3.5-cm dishes, pipette 1.5 ml cold HBSS to each dish and place them on ice packs.
- For each rat pup to be used, place a 1 ml aliquot of 0.25% trypsin-EDTA in a 37 °C water bath to prewarm it.
- Coat a 3.5-cm dish with 1 ml PLL solution (0.1 mg/ml in distilled water, see Recipes) at 37 °C in a CO2 incubator overnight.
- Tissue collection
- Anaesthetize the neonatal rat (postnatal 2-4 days) by cooling it on ice until it stops moving.
Note: Animal use was approved by the Southern Medical University Animal Care and Use Committee in accordance with the guidelines for the ethical treatment of animals. All efforts were made to minimize animal sacrifice and suffering. - Sterilize the rat with 75% ethanol.
- Rapidly decapitate the rat with big surgical scissors and forceps, dispose the head into a collection bag.
- Place the rat on the dissecting board in prone posture (dorsal side up), and pin the extremities with needles (see Figure 2A).
- Using a set of small scissors and forceps, cut the back skin along the median line from rostral to caudal, separate the skin from the body from medial to lateral (see Figure 2B).
- Using the spring scissors and fine forceps, separate the shoulder blades from backbone, and pin the shoulder blades on the dissecting board.
- Using the spring scissors and fine forceps, carefully expose the brachial plexuses and dissociate the connective tissue from the nerves. Then cut the nerves at the distal end (see Figures 2C and 2D).
- Using the spring scissors and fine forceps, carefully separate the hamstring muscles from the knee to the hipbone to expose the sciatic nerves. Then cut the sciatic nerves at the distal end (see Figure 2E).
Note: Do not cut the proximal end of the nerves in steps B7 and B8. The nerves are not directly dissected out in these steps. Instead, the nerves should be dragged out from intervertebral foramina as described in the following step B11. - Using the spring scissors and fine forceps, cut the bilateral vertebral arch and remove the vertebral laminae from rostral to caudal to expose the spinal cord (see Figure 2F).
- Using the spring scissors and fine forceps, remove the spinal cord carefully to expose the underlying dorsal root ganglia (see Figures 2G and 2H).
- Using the fine forceps, grasp the ganglion carefully and then drag out the connecting spinal nerve and put it in a 3.5-cm dish with ice-cold HBSS. Repeat this procedure to harvest all of the spinal nerves from caudal to rostral (mainly including brachial plexus nerves, intercostal nerves and sciatic nerves) (see Figures 2I and 2J).
Notes:- By grasping the ganglia in the inner of canalis vertebralis (spinal canal), all of the spinal nerves can be easily found and collected.
- Slowly and gently drag the spinal nerves out through the intervertebral foramina will help to obtain the nerves longer.
- Harvesting of the nerves is most easily done under a stereomicroscope. It is highly suggested.
- By grasping the ganglia in the inner of canalis vertebralis (spinal canal), all of the spinal nerves can be easily found and collected.
- Use the superfine forceps to remove the connective tissue that adheres to the nerve, and cut off the ganglion with fine scissors. Transfer the clean nerve into another 3.5-cm dish with ice-cold HBSS. Repeat this procedure to clear all of the nerves (see Figures 2K and 2L).
Notes:- About 30 nerves including sciatic nerves, branches of brachial plexuses and intercostal nerves can be obtained from each rat pup.
- Each collected nerve is with a ganglion in the proximal end. The ganglion looks like a head of the nerve and more transparent than the nerve (see Figure 2K, arrows). Meanwhile, rare connective tissue which appears as membranaceous tissue with blood vessels may adhere to the nerves (see Figure 2K, arrowheads).
- Nerves harvested by dragging out through intervertebral foramina are always free from epineuria because the epineuria adhere tightly to the intervertebral foramina. This is a major advantage of this protocol. As traditional methods inevitably bring in the epineuria which tightly wrap the nerves (Weinstein et al., 2001; Tao, 2013). And the epineuria are the main source of contaminating cells (such as fibroblasts) in the SC cultures.
- Another advantage of the present protocol is that a large number of nerves can be obtained from each rat pup, rather than just two sciatic nerves in traditional methods. Thus, the sacrificed animals can be significantly reduced.
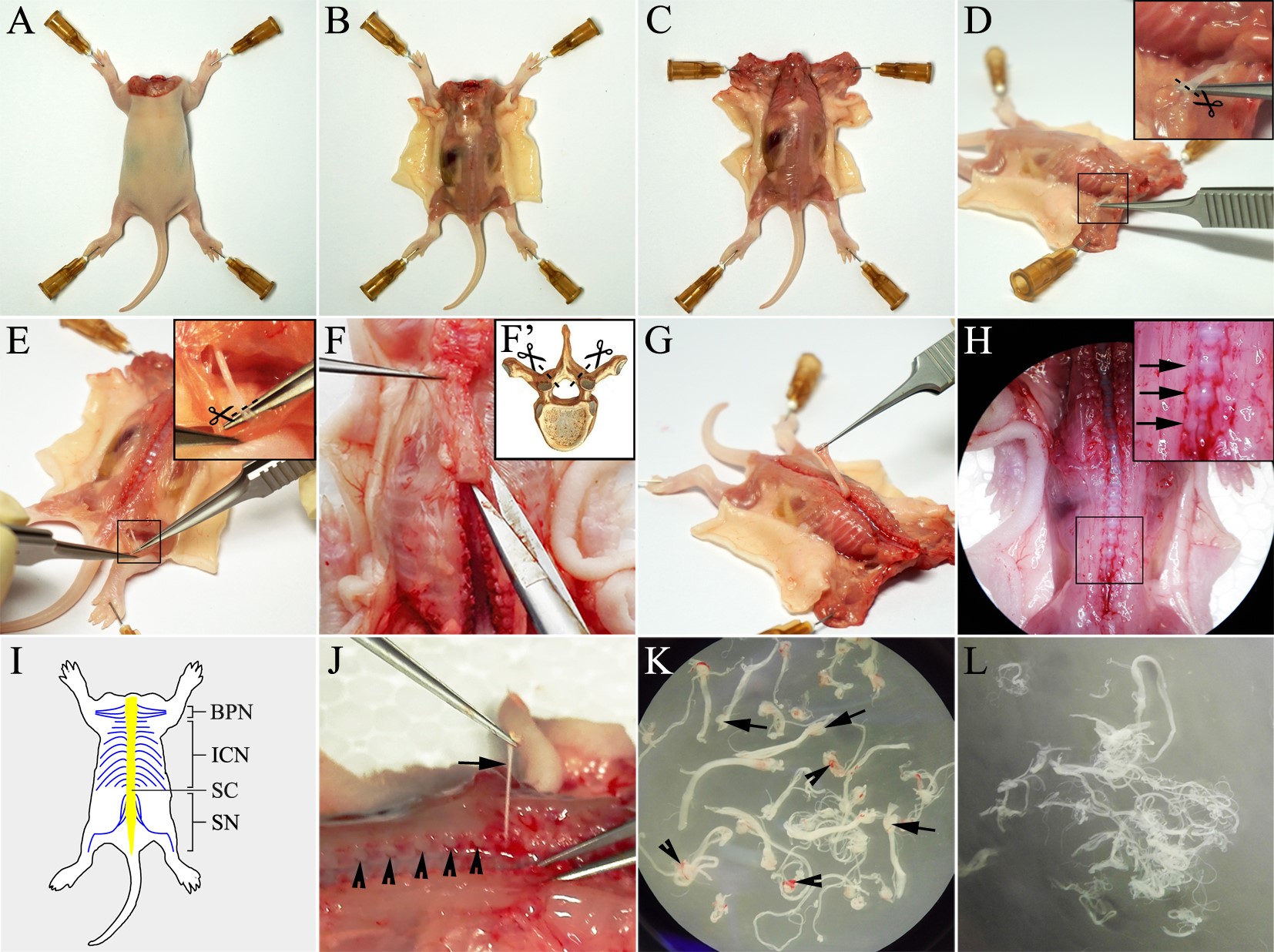
Figure 2. Step-by-step procedures of harvesting spinal nerves. A. Decapitated rat pup is pinned on the dissecting board. B. The back skin is cut and separated from the body. C. The shoulder blades are separated from the backbone and pinned on the dissecting board. D. The brachial plexuses are exposed and its distal ends are cut. E. The sciatic nerve is exposed and its distal ends are cut. F. Open vertebral canal by laminectomy using a pair of spring scissors. G. The spinal cord is dragged and removed from rostral to caudal. H. The dorsal root ganglia are exposed (arrows) after spinal cord is removed off. I. Schematic diagram shows the spinal nerves (in blue) mainly including brachial plexus nerves (BPN), intercostal nerves (ICN) and sciatic nerves (SN) available for harvesting. Spinal cord (SC) is indicated in yellow. J. A representative image shows the nerve is dragged out through an intervertebral foramen (arrow) while some dorsal root ganglia remain in the foramina (arrowheads). K. The collected nerves are with ganglia (arrows) and sometimes with connective tissue (arrowheads). L. The nerves after the ganglia and connective tissue are removed off.
- About 30 nerves including sciatic nerves, branches of brachial plexuses and intercostal nerves can be obtained from each rat pup.
- Anaesthetize the neonatal rat (postnatal 2-4 days) by cooling it on ice until it stops moving.
- Dissociation and cell seeding
- Transfer the clean nerves to a 1.5 ml tube and cut the nerves into small pieces of 0.5-1 mm long.
- Add 1 ml of prewarmed 0.25% trypsin-EDTA into the tube. Cap the tube and incubate it in a 37 °C water bath for 30 min with intermittent vibration every 10 min.
- Terminate the trypsinization by supplementing with 100 μl FBS into the medium. And then gently triturate the sample 30-35 times using a 1 ml pipette tip to make single cell suspension.
Note: Triturate gently to avoid bubble formation and splashing. After 30-35 times of pipetting up and down, some residual clumps remain in the cell suspension. - Filter the cell suspension through a 100 μm cell strainer to remove the residual clumps.
Note: The clumps are mainly axonal debris and un-separated tissues. - Centrifuge the cell suspension at 106 x g (Rcf) for 10 min at room temperature (RT), and discard the supernatant.
- Resuspend the cell pellet in 300 μl 10% FBS in DMEM/F12 (see Recipes). Plant the cell suspension in the middle of the dried PLL-coated 3.5-cm dish. Do not shake the dish lest the cell suspension spread out to adhere to the wall of the dish. Culture the cells in a CO2 incubator for 2 h to facilitate cell attachment.
Notes:- A total number of ~1 x 106 cells can be obtained from each rat for one dish in our routine practice.
- Minimize the volume of plating medium (300 μl 10% FBS) can increase the cell density and facilitate cell attachment to the dish.
- A total number of ~1 x 106 cells can be obtained from each rat for one dish in our routine practice.
- Add 1 ml 10% FBS to the dish after the cells attach to the dish.
- Transfer the clean nerves to a 1.5 ml tube and cut the nerves into small pieces of 0.5-1 mm long.
- Purification and expansion
- In the next day, replace the culture medium with DMEM/F12 containing 10% FBS and 10 μM AraC (see Recipes) to eliminate contaminating fibroblasts.
Note: In the absence of mitogenic factors, such as heregulin and forskolin, SCs proliferate quite slowly while fibroblasts do proliferate quickly. AraC is an antimetabolic agent that impairs DNA synthesis and kills dividing cells. Therefore, fibroblasts are eliminated efficiently while SCs survive well in an appropriate treating time span (48 h). - After 48 h, replace the medium with SC culture medium (see Recipes) to expand SCs.
- When the culture reaches 90% confluence, cells are routinely passaged at a ratio of 1:3 to expand the cells. And cells from the 3-5th passages are used for experiments in our lab.
Note: Primary cultured SCs of the 3-5th passages show bipolar and tripolar shapes (see Figure 3A). The purity of these cells can reach 98% identified by immunostaining of SC specific markers (see Figures 3B-3D). We found that SCs proliferate much slower after they were passaged for more than 5 times. In addition, most published papers about SC biology stated that primary SCs from neonatal rat were used in the 3-5th passages. So, in our lab, all SCs used for studies are from the 3-5th passages.
- In the next day, replace the culture medium with DMEM/F12 containing 10% FBS and 10 μM AraC (see Recipes) to eliminate contaminating fibroblasts.
Data analysis
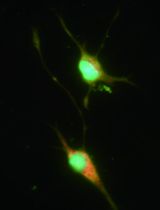
To identify the purity of SCs in our cultures, GFAP, P75 and S100 were used as the specific markers of SCs to perform immunostaining as our previous report (Wen et al., 2017). Briefly, cultured cells were fixed with 4% PFA (see Recipes) for 20 min. Then cells were penetrated with 0.1% Triton X-100 (see Recipes) for 30 min and incubated with blocking buffer (see Recipes) at RT for 1 h. And then the cells were incubated with primary antibodies (1:400 diluted in blocking buffer) at 4 °C overnight, followed by an incubation with Alexa-488 fluorescent conjugated secondary antibodies (1:400 diluted in blocking buffer) at RT for 2 h. After immunostaining, cells are incubated with 1 μg/ml DAPI (see Recipes) for 5 min at RT to stain nuclei. As shown in Figure 3, purified SCs show bipolar or tripolar shapes, and are positive for specific markers including S100, GFAP, and P75.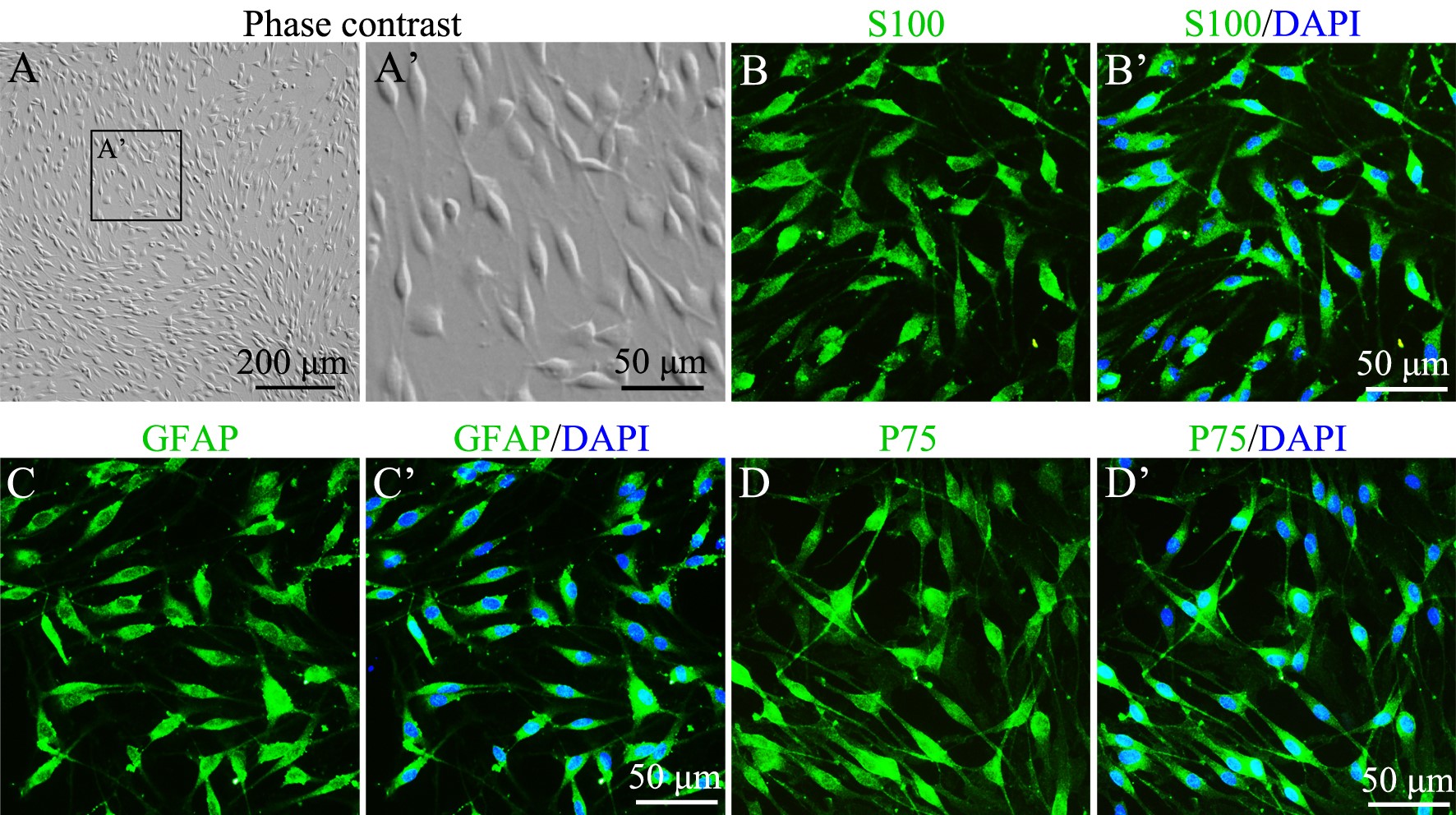
Figure 3. Identification of cultured Schwann cells. A and A’. Primary cultured SCs under phase contrast microscope show bipolar or tripolar shapes. B-D’. Primary SCs are identified by immunostaining with specific markers including S100 (B, B’), GFAP (C, C’) and P75 (D, D’).
Recipes
- 100x poly-L-lysine hydrobromide (PLL)
Dissolve 500 mg PLL powder in 50 ml distilled water to make 100x stock solution of 10 mg/ml - 10% fetal bovine serum (10% FBS) DMEM/F12 media
Dilute 5 ml FBS and 500 μl Pen/Strep into 44.5 ml Dulbecco’s modification of Eagle’s medium/Ham’s F-12 50/50 Mix (DMEM/F12) - 1,000x cytosine arabinoside (AraC)
Dissolve 4.86 mg AraC in 2 ml distilled water to make 1,000x stock solution of 10 mM and sterilize the solution by filtration
Store at -20 °C and protect from light - 5,000x heregulin
Dissolve 50 μg heregulin in 1 ml sterile 0.1% bovine serum albumin (BSA) to make 5,000x stock solution of 50 ng/μl - 10,000x forskolin
Dissolve 10 mg forskolin in 812 μl DMSO to make 10,000x stock solution of 30 mM - SC culture medium
DMEM/F12 containing 3% FBS supplement with 10 ng/ml heregulin, 3 μM forskolin, and 1% Pen/Strep
Dilute 1.5 ml FBS, 10 μl 5,000x Heregulin, 5 μl 10,000x Forskolin and 500 μl Pen/Strep into 48 ml DMEM/F12 - 4% paraformaldehyde (PFA)
Dilute 4 g PFA in 100 ml PBS
Stir at 65 °C until complete dissolution, and store at 4 °C - 0.1% Triton X-100
Dilute 100 μl Triton X-100 into 100 ml PBS - Blocking buffer
Dissolve 0.5 g gelatin (5%, w/v) in 10 ml PBS, and add 300 μl Triton X-100 (0.3%) to the buffer - 1,000x 4, 6-diamidino-2-phenylindole (DAPI)
Dissolve 5 mg DAPI in 5 ml distilled water to make 1,000x stock solution of 1 mg/ml, dilute it to 1x with PBS before used
Acknowledgments
This protocol is adapted from the previously published paper (Wen et al., 2017). This work was supported by the National Key Basic Research Program of China (2014CB542202 and 2014CB542205), National Natural Science Foundation of China (30973095, 81371354 & 81571182); Science and Technology Project of Guangzhou (12C32121609) and Natural Science Foundation of Guangdong Province (S2013010014697) to J Guo.
References
- Assouline, J. G., Bosch, E. P. and Lim, R. (1983). Purification of rat Schwann cells from cultures of peripheral nerve: an immunoselective method using surfaces coated with anti-immunoglobulin antibodies. Brain Res 277(2): 389-392.
- Brockes, J. P., Fields, K. L. and Raff, M. C. (1979). Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res 165(1): 105-118.
- Haastert, K., Mauritz, C., Chaturvedi, S. and Grothe, C. (2007). Human and rat adult Schwann cell cultures: fast and efficient enrichment and highly effective non-viral transfection protocol. Nat Protoc 2(1): 99-104.
- Jin, Y. Q., Liu, W., Hong, T. H. and Cao, Y. (2008). Efficient Schwann cell purification by differential cell detachment using multiplex collagenase treatment. J Neurosci Methods 170(1): 140-148.
- Oda, Y., Okada, Y., Katsuda, S., Ikeda, K. and Nakanishi, I. (1989). A simple method for the Schwann cell preparation from newborn rat sciatic nerves. J Neurosci Methods 28(3): 163-169.
- Pannunzio, M. E., Jou, I. M., Long, A., Wind, T. C., Beck, G. and Balian, G. (2005). A new method of selecting Schwann cells from adult mouse sciatic nerve. J Neurosci Methods 149(1): 74-81.
- Tao, Y. (2013). Isolation and culture of Schwann cells. Methods Mol Biol 1018: 93-104.
- Vroemen, M. and Weidner, N. (2003). Purification of Schwann cells by selection of p75 low affinity nerve growth factor receptor expressing cells from adult peripheral nerve. J Neurosci Methods 124(2): 135-143.
- Weinstein, D. E. and Wu, R. (2001). Isolation and purification of primary Schwann cells. Curr Protoc Neurosci Chapter 3: Unit 3.17.
- Wen, J., Qian, C., Pan, M., Wang, X., Li, Y., Lu, Y., Zhou, Z., Yan, Q., Li, L., Liu, Z., Wu, W. and Guo, J. (2017). Lentivirus-mediated RNA interference targeting RhoA slacks the migration, proliferation, and myelin formation of Schwann cells. Mol Neurobiol 54(2): 1229-1239.
- Wood, P. M. (1976). Separation of functional Schwann cells and neurons from normal peripheral nerve tissue. Brain Res 115(3): 361-375.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wen, J., Tan, D., Li, L. and Guo, J. (2017). Isolation and Purification of Schwann Cells from Spinal Nerves of Neonatal Rat. Bio-protocol 7(20): e2588. DOI: 10.21769/BioProtoc.2588.
Category
Neuroscience > Peripheral nervous system > Schwann cell
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.