- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Assays for Eukaryotic Leading/Lagging Strand DNA Replication
Published: Vol 7, Iss 18, Sep 20, 2017 DOI: 10.21769/BioProtoc.2548 Views: 10271
Reviewed by: Gal HaimovichVamseedhar RayaproluDavid Paul
Original research article
The authors used this protocol in:

Jan 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The eukaryotic replisome is a multiprotein complex that duplicates DNA. The replisome is sculpted to couple continuous leading strand synthesis with discontinuous lagging strand synthesis, primarily carried out by DNA polymerases ε and δ, respectively, along with helicases, polymerase α-primase, DNA sliding clamps, clamp loaders and many other proteins. We have previously established the mechanisms by which the polymerases ε and δ are targeted to their ‘correct’ strands, as well as quality control mechanisms that evict polymerases when they associate with an ‘incorrect’ strand. Here, we provide a practical guide to differentially assay leading and lagging strand replication in vitro using pure proteins.
Keywords: Eukaryotic DNA replicationBackground
Using pure proteins from Saccharomyces cerevisiae, our lab was the first to reconstitute a functional eukaryotic DNA replisome, a ~2 MDa complex that includes the 11-subunit CMG helicase (complex of Cdc45, Mcm2-7, GINS heterotetramer), the 4-subunit DNA polymerase (Pol) ε, the 4-subunit Pol α-primase, the PCNA (Proliferating Cell Nuclear Antigen) clamp homotrimer ring shaped processivity factor that encircles duplex DNA, the 5-subunit clamp loader RFC (Replication Factor C) that uses ATP to open and close the PCNA sliding clamp ring onto primed sites for polymerase processivity, and the RPA (Replication Protein A) heterotrimeric single-strand DNA binding protein that removes secondary structure obstacles to DNA polymerase progression. In our initial studies we discovered that Pol ε is targeted to CMG on the leading strand after priming by Pol α-primase, while Pol δ is targeted to PCNA clamps on the lagging strand primed sites (Georgescu et al., 2014; Langston et al., 2014). We next reconstituted a functional coupled leading/lagging strand replisome which included the 4-subunit Pol α-primase and 3-subunit Pol δ, in which we demonstrated that Pol ε is inactive on the lagging strand and Pol ε is inactive on the leading strand (Georgescu et al., 2015). Interestingly, the Pol α-primase, which lacks proofreading activity, was active with CMG on both strands, but when either Pol ε or Pol δ are present, which both contain a proofreading 3’-5’ exonuclease for high fidelity synthesis, they take over from the low fidelity Pol α-primase on either strand. However, Pol ε and Pol δ only performed optimal synthesis when on their respective correct strands (Georgescu et al., 2015). In a subsequent study we characterized the unprecedented quality control mechanisms that exclude these polymerases from incorrect strands, a job that bacterial replisomes do not need to do because they utilize identical polymerases for both strands (Schauer et al., 2017). We found that on the lagging strand, Pol ε is excluded from primed sites by competition with the RFC clamp loader for the primer terminus, while CMG binds and protects Pol ε from RFC inhibition on the leading strand. In contrast Pol δ is preferentially targeted to PCNA on lagging strand primed sites through a tight binding affinity to PCNA clamps that is over 20-fold greater than the PCNA affinity to Pol ε and is unaffected by competition by the RFC clamp loader (Schauer et al., 2017). Interestingly, no stabilizing interaction with CMG exists for Pol δ (Schauer et al., 2017). Furthermore, Pol δ is less stable on a completed DNA than when idling at a primer terminus or extending a primer. Specifically, Pol δ is known to be stable for over a half hour with PCNA, consistent with its high processivity, but upon completing replication of a section of DNA, and bumping into a completed dsDNA region, it dissociates rapidly (i.e., < 1 min) from PCNA-DNA in a process referred to as collision release (Langston and O’Donnell, 2008; Langston et al., 2014).This inherent instability of Pol δ-PCNA upon completing replication may serve as a quality control to destabilize Pol δ-PCNA on the leading strand because Pol δ-PCNA is much faster than CMG unwinding and will be in a constant state of having completed DNA and collision with CMG (Schauer et al., 2017). Destabilization of Pol δ-PCNA when there is no more DNA to be extended should not be taken to mean that Pol δ instantly ejects from PCNA. For example, Pol δ-PCNA remains on DNA for a few seconds to fill-in short gaps upon RNA removal at 5’ ends of Okazaki fragments (Stodola and Burgers, 2016).
In interrogating these various activities, we observed that CMG does not load onto small (100-200 bp) rolling circle replication substrates, which are often used to study replisome behavior in bacterial systems. Thus, we turned to linear DNA fork assays as an alternative to address biochemical mechanisms in eukaryotic replication. These assays enable one to easily separate leading from lagging strand replication activity by synthesis of a long linear DNA that has no dC in one strand, and thus no dG in the other strand. By doing so, one can specifically monitor leading or lagging strand synthesis depending on the radioactive deoxyribonucleoside triphosphate (dNTP) used in the assay.
Materials and Reagents
- Razor blade
- 1.57 mm OD polyethylene tubing (e.g., Clay Adams® Intramedic®, BD, catalog number: 427431 )
- Sephadex microcentrifuge columns (Illustra Microspin G-25) (GE Healthcare, catalog number: 27-5325-01 )
- Plastic wrap (e.g., Fisherbrand Clear Plastic Wrap, Fisher Scientific, catalog number: 22-305654 )
- C-fold paper towels (e.g., Scott paper towels, KCWW, Kimberly-Clark, catalog number: 01510 )
- Positively charged nylon DNA blotting membrane (Hybond-N+, 30.0 x 50.0 cm) (GE Healthcare, catalog number: RPN3050B )
- Chromatography transfer paper (Whatman 3MM, 46.0 x 57.0 cm) (GE Healthcare, catalog number: 3030-917 )
- Syringe tip (e.g., B-D 18 G 1 ½ PrecisionGlide® Needle) (BD, catalog number: 305196 )
- phiX174 virion DNA, 1 mg/ml (New England Biolabs, catalog number: N3023L )
- Phi29 DNA polymerase (New England Biolabs, catalog number: M0269S )
- 100 mM dNTP (deoxynucleotide triphosphate) set (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0181 )
- 1 μM CMG (Cdc45 Mcm2-7 Gins) helicase (see Georgescu et al. [2014] for purification details)
- pUC19, 1 mg/ml (New England Biolabs, catalog number: N3041L )
- BsaI-HF with CutSmart buffer (New England Biolabs, catalog number: R3535L )
- ‘blockLd’ oligo*
- ‘blockLg’ oligo*
- ‘Pr1B’ oligo*
- ‘160Ld’ oligo*
- ‘91Lg’ oligo*
- ‘Fork primer’ oligo*
- Nucleotide-biased template (synthesized by Biomatik, Wilmington DE)*
*Note: See Supplementary file 1.
- T4 ligase, including 10x ligase buffer (New England Biolabs, catalog number: M0202M )
- 100 mM ATP (GE Healthcare, catalog number: 27-2056-01 )
- 0.5 M EDTA, disodium salt (Sigma-Aldrich, catalog number: E5134 )
- 5 M NaCl (Sigma-Aldrich, catalog number: S9888 )
- Sepharose 4B size exclusion chromatography resin (GE Healthcare, catalog number: 17012001 )
- 1 kb MW marker (New England Biolabs, catalog number: N3232L )
- Ethidium bromide (EthBr, 10 mg/ml) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15585011 )
- T4 kinase and 10x T4 kinase buffer (New England Biolabs, catalog number: M0201L )
- 32P-γ-ATP, 3,000 Ci/mmol, 3.3 μM (PerkinElmer, catalog number: BLU002A )
- Type XI low-melt agarose (Sigma-Aldrich, catalog number: A3038 )
Note: This product has been discontinued. - BtsCI (New England Biolabs, catalog number: R0647L )
- β-Agarase I (New England Biolabs, catalog number: M0392L )
- 3 M sodium acetate (CH3COONa), pH 5.2 (Sigma-Aldrich, catalog number: S2889 )
- Isopropanol (Sigma-Aldrich, catalog number: 190764 )
- Glycogen, molecular biology grade (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0561 )
- Ethanol (Sigma-Aldrich, catalog number: E7023 )
- 1 μM RFC (Replication Factor C; see Georgescu et al. [2014] for purification details)
- 5 μM PCNA (Proliferating Cellular Nuclear Antigen; see Georgescu et al. [2014] for purification details)
- 2 μM Pol ε (see Georgescu et al. [2014] for purification details)
- 2 μM Pol δ (see Georgescu et al. [2014] for purification details)
- 2 μM Pol α (see Georgescu et al. [2014] for purification details)
- 20 μM RPA (Replication Protein A; see Georgescu et al. [2014] for purification details)
- 32P-α-dCTP, 3,000 Ci/mmol, 3.3 μM (PerkinElmer, catalog number: BLU013H )
- 32P-α-dGTP, 3,000 Ci/mmol, 3.3 μM (PerkinElmer, catalog number: BLU514H )
- LE agarose (BioExpress, GeneMate, catalog number: E-3120-500 )
- 10 N sodium hydroxide (NaOH) (Fisher Scientific, catalog number: SS255 )
- Glycerol
- Xylene cylanol
- Tris-HCl, pH 8.0
- Tris base (RPI, catalog number: T60040-500.0 )
- Boric acid (RPI, catalog number: B32050-5000.0 )
- Sodium citrate
- 1-Butanol
- Tris-acetate, pH 7.5
- Bovine serum albumin (BSA) (New England Biolabs, catalog number: B9000S )
- Tris(2-carboxyethyl)phosphine (TCEP) pH 7.5
- 100 mM dithiothreitol (DTT) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0861 )
- Potassium glutamate
- Magnesium acetate
- 1% SDS
- 6x gel loading dye (see Recipes)
- TE buffer, pH 8.0 (see Recipes)
- 10x TBE (Tris/Borate/EDTA; see Recipes)
- DNA elution buffer (see Recipes)
- 20x SSC (see Recipes)
- 1-Butanol saturated water (see Recipes)
- 5x TDBG (see Recipes)
- 10x MK (see Recipes)
- dA/dC mix (see Recipes)
- dT/dG mix (see Recipes)
- T/G/C mix (see Recipes)
- Stop solution (see Recipes)
- Alkaline running buffer (see Recipes)
Equipment
- Heating block (e.g., VWR, catalog number: 12621-084 )
- Fraction collector (e.g., Gilson, model: F203B )
- Variable mode gel imager (e.g., GE Typhoon)
- UV-vis spectrophotometer (e.g., Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000 )
- Vacuum dessicator (e.g., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 5309-0250 )
- UV light box
- UV blocking face shield (e.g., Sigma-Aldrich, catalog number: F8142 )
Note: This product has been discontinued. - Microcentrifuge
- Conductivity meter (e.g., Radiometer Medical, model: CDM 80 )
- Temperature controlled water bath with microcentrifuge tray (e.g., LabX, model: Lauda E100 and Brinkman 30x x 1.5 ml)
Manufacturer: LAUDA-Brinkmann, model: E100 . - Phosphorimaging screen (GE Healthcare)
- Phosphor imager (e.g., GE Typhoon)
- A heavy weight
Note: We use giant lead blocks that we found; a ~50 lb dumbell would work. - Computer with ImageJ and spreadsheet software (e.g., Apache Open Office) installed
- 1 x 30 cm glass column (e.g., glass Econo-Columns®) (Bio-Rad Laboratories, catalog number: 7371032 )
- 100 ml agarose gel electrophoresis apparatus
- Styrofoam box large enough to fit 100 ml agarose gel electrophoresis apparatus
- Electrophoresis power supply (e.g., Pharmacia Biotech, catalog number: EPS 3500 XL )
- Analytical balance
- Protective plexiglass samples shield
- 20 x 14 cm horizontal agarose gel electrophoresis apparatus (C.B.S. Scientific, catalog number: SGU-014T-02 )
Software
Procedure
- Linear forked DNA template construction
We primarily use two types of linear forked DNA substrates to assay leading/lagging strand replication: a linear pUC based natural sequence substrate (referred to below as 3kbf, for 3 kb fork), and a 3.2 kb nucleotide-biased, leading/lagging strand substrate that is generated synthetically such that one strand has no dC and the other strand has no dG (referred to as 3nbf, for 3 kb nucleotide biased fork). The 3nbf substrate can be used in leading and lagging strand assays to monitor each strand independent of the other (see Figure 1). We sometimes prefer to use a 5’-32P-labeled primer to quantify leading strand replication as it is a direct reporter of primer elongation compared to use of 32P-dNTPs because it lacks potential end-labeling artifacts. Though both substrates can accommodate a primer, we typically only use the 3kbf substrate for this purpose because it is much simpler to prepare (i.e., does not require purification of the 3 kb synthetic nucleotide biased section from a low melt gel). Although the template length and composition differs between these substrates, we use the same small synthetic fork construct that is ligated onto either of these substrates.
The fork is composed of a 160 mer for the leading strand and a 91 mer for the lagging strand. The leading strand contains a 3’ (dT)34 tail for CMG loading, a 37 nucleotide primer annealing site followed by a 4 nucleotide region lacking dC and dA residues and a 10 nucleotide region lacking dC and dT residues (see Recipes for oligonucleotide composition). The 4 nucleotide region upstream of the primer allows ‘idling’ of Pol ε on the leading strand by only including dATP and dCTP (i.e., to prevent the 3’-5’ exonuclease activity of Pol ε from degrading the primer). The 10 nucleotide region prevents ‘runaway’ Pol ε after potential misincorporation (i.e., even with dTTP withheld). Following these sequences, we include a (dT)40 spacer to the fork junction designed to accommodate the footprint of CMG (~10 nm). The lagging strand contains a (dT)60 5’-tail and 35 bp complementary to the 5’ end of the leading strand. The leading strand fork template oligo typically contains four 3’ phosphorothioate bonds to protect against exonuclease activity inherent in DNA polymerases by substituting a sulfur atom for a non-bridging phosphate oxygen atom in the oligonucleotide backbone (available as a modification from DNA synthesis companies, such as IDT). See Figure 1 for a schematic of the nucleotide biased substrate.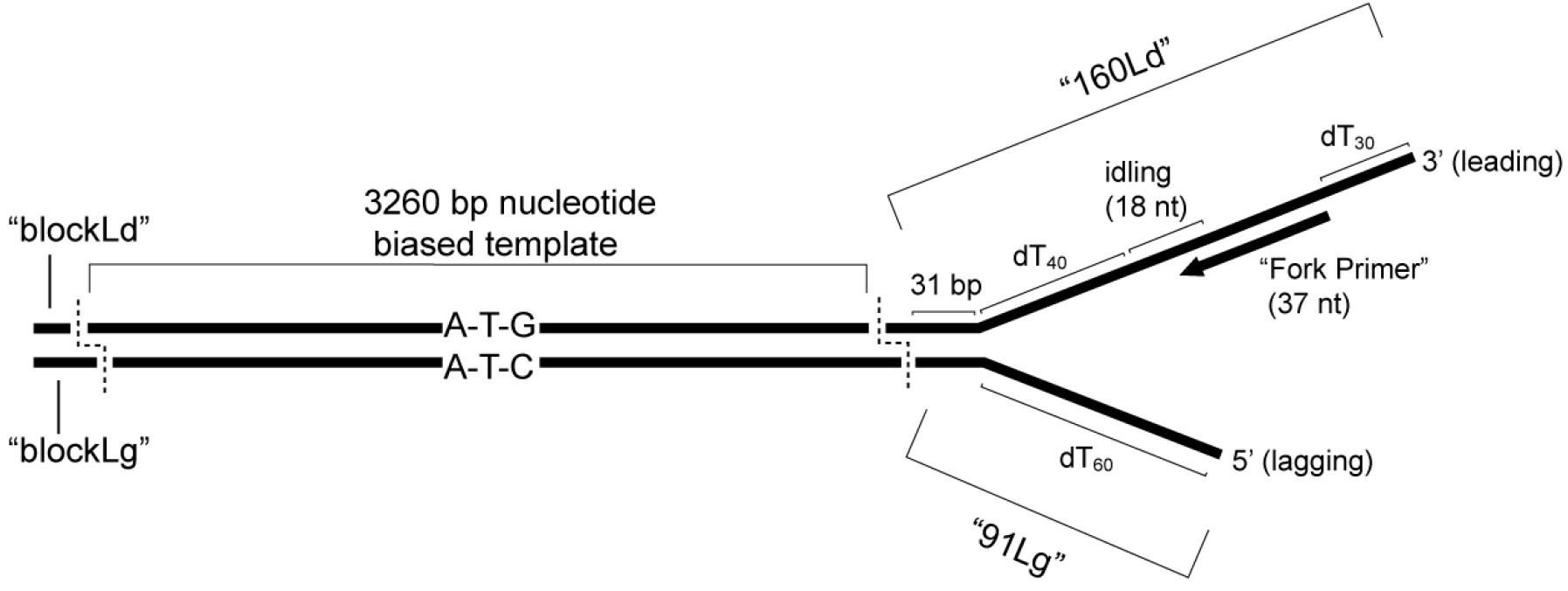
Figure 1. Schematic of nucleotide biased substrate. The “3nbf” substrate, including the oligonucleotides described in the text, is shown with the relevant features highlighted. The bias is designed such that 32P-α-dCTP will only be incorporated during leading strand synthesis, while 32P-α-dGTP will only be incorporated during lagging strand synthesis. Dotted lines indicate restriction enzyme cut sites discussed in the text. Note that the “3kbf” substrate, which is constructed using the same synthetic fork but using a 2,686 bp template with natural (unbiased) sequence, is not shown.
3kbf substrate construction (typically used for leading strand reactions using a 32P-primer)- Resuspend ‘160Ld’ to 20 μM in TE buffer (see Recipes)
- Resuspend ‘91Lg’ to 500 μM in TE buffer.
- Digest 35 pmol pUC19 with 4 μl BsaI-HF (20 U/μl) in 1x CutSmart buffer in 40 μl total at 37 °C for 4 h.
- Heat inactivate BsaI-HF at 75 °C for 30 sec.
- Add 17.5 μl (350 pmol) 160Ld and 4.2 μl (2,100 pmol) 91Lg in TE, and add 238.3 μl TE to 400 μl.
Note: The 10:1 excess of fork to template is required to prevent concatamerization of linearized pUC19, and the 6:1 excess of 91Lg to 160Ld is to ensure all ligated 160Ld is annealed to 91Lg. - Anneal fork to template by heating to 95 °C for 5 sec and let heat block cool to RT on benchtop.
- Ligate fork onto linear pUC19 by adding 15 μl T4 ligase, 36 μl 10x CutSmart buffer, 4 μl 100 mM ATP (1 mM final), adding TE to 400 μl volume. Incubate at 16 °C for ≥ 16 h.
- Add 10 μl 0.5 M EDTA to chelate Mg2+.
- Heat inactivate T4 ligase at 65 °C for 20 sec.
- Add 10 μl 5 M NaCl to bring [NaCl] to > 100 mM (necessary to prevent nonspecific binding to Sepharose 4B column).
- Pour a ~20 ml Sepharose 4B column and equilibrate with > 2 column volumes (CV) of cold DNA elution buffer (see Recipes).
- Adapt syringe tip to end of the column. Adapt polyethylene tubing to syringe tip and couple to fraction collector with the minimum length required for the device to reach all tubes.
- Add 420 μl reaction to 20 ml Sepharose 4B column, ensuring that buffer has completely seeped into the column bed before loading the reaction volume.
- After the sample has completely penetrated the column bed, add and elute with cold DNA elution buffer.
- Collect ~227 μl fractions (10 drops/fxn for the tubing we use). Under these conditions, the template should elute around fraction 20.
- Run 7 μl of each fraction on a 1% TBE agarose gel alongside a 1 kb MW marker (with gel loading buffer) at 100 V until the bromophenol blue marker reaches 2/3 the way down.
- Stain with 2-5 μl EthBr in 1x TBE and visualize on a gel imager.
Notes:- The gel shift of the linearized plasmid from ligated fork DNA is very small, so do not expect to see a difference. However the ligated fork DNA will elute much earlier than the excess of free synthetic forked DNA, which can be visualized on a properly stained/destained gel.
- We have quantified fork ligation with radiolabeled fork, and it is highly (> 95%) efficient, thus the 3 kb template is attached to the DNA fork under the conditions described above.
- Nonetheless, we encourage users to actually monitor their ligation efficiency using 32P-labelled fork DNA to be sure of ligation, and use of phi29 DNA polymerase in the extension of a 32P-labeled primer (i.e., see below) to further ensure ligation efficiency.
- The gel shift of the linearized plasmid from ligated fork DNA is very small, so do not expect to see a difference. However the ligated fork DNA will elute much earlier than the excess of free synthetic forked DNA, which can be visualized on a properly stained/destained gel.
- Save fractions containing the ~2.7 kb band (this will be the sole visible band amongst all the fractions) and measure DNA concentration via A260 using a UV/Vis spectrophotometer.
- Follow manufacturer’s protocols to 5’-end-label the primer with T4 kinase and 32P-γ-ATP. We typically use a 25 μl reaction with a final primer concentration of 400 nM. Purify away free 32P-γ-ATP with a G-25 column that has been equilibrated with TE, according to the manufacturer’s protocol.
- To prime the 3kbf substrate with 32P-primer, add the required amount of 3kbf substrate with an equimolar amount of 32P-primer. Minimize the total volume, add 20x SSC (see Recipes) to a final 1x concentration, heat to 95 °C and cool to RT on the benchtop.
3nbf substrate construction- Resuspend ‘160Ld’ to 20 μM in TE buffer.
- Resuspend ‘91Lg’ to 500 μM in TE buffer.
- Digest ~400 μg plasmid containing 3.2 kb nucleotide-biased template (see Supplementary file 1 for sequence) with 12 μl (20 U/μl) BsaI-HF in 1x CutSmart buffer in 400 μl total at 37 °C for 7 h.
- Before the previous step is finished, run 0.5 μl on a 0.8% TBE agarose gel at 100 V until the bromophenol blue reaches 2/3 the way down to ensure complete cutting. If cutting is not complete, add more BsaI-HF and digest more; repeat until cutting is complete.
- Add 12 μl BtsCI (20 U/μl) plus enough 10x CutSmart buffer and to compensate for added volume. Incubate at 50 °C for 6 h.
- Before digest is finished, run 0.5 μl on gel as in step 4 (Procedure A, 3nbf substrate construction) to ensure complete digest.
- Add 21.5 μl 0.5 M EDTA.
- Place sample in vacuum desiccator for 6-8 h to reduce volume by roughly 1/3.
- Run entire sample on 1.2% TBE low-melt agarose gel (well is 8 of 15 teeth of comb taped together). Add 2 μl 10 mg/ml EthBr to 100 ml gel. Pre-chill TBE, adding 2 μl 10 mg/ml EthBr for every 100 ml running buffer used. Run at 40-50 V with the apparatus sitting in a Styrofoam box full of ice-water (a cold room is not sufficient) until the bromophenol blue reaches the bottom third of the gel. Donning UV-protective face shield, visualize on a UV light box and cut out the largest (3,260 bp) band out with a razor blade. Weigh the gel slice for the next steps.
- Melt gel slices at 65 °C.
- Weigh melted slices and add 1/9 V Tris-HCl, pH 6.8 and 1/224 V 0.5 M EDTA, under the rough approximation that the density of the gel is 1 g/ml.
- Cool to 42 °C.
- Add ~5 μl β-agarase I per gram of gel. Incubate at 42 °C for 5-7 h.
- Repeat step 13 (Procedure A, 3nbf substrate construction).
- Isopropanol precipitate the DNA fragment.
Add 1/10 the volume of 3 M sodium acetate, pH 5.2, followed by an equal volume of isopropanol. Incubate for 1 h at -20 °C. Spin in a microcentrifuge at ≥ 15,000 x g for 30 min.
Note: If too much agar has not been digested sufficiently to small oligosaccharides, you will see a lot of white precipitate. You will need to finish the precipitation, re-suspend in aqueous solution, and go back to step 13 (Procedure A, 3nbf substrate construction) to perform a further β-agarase I digestion. - Decant supernatant and resuspend pellet in TE pH 8.0.
- Remove EthBr by extraction with an equal volume of 1-butanol saturated water (see Recipes). Gently vortex to mix. Spin at 1,500 x g, saving the bottom solution. Continue extracting until there is no visible pink color. Extract 3 more times.
- Ethanol precipitate
Add 1 μl glycogen (20 mg/ml), 1/10 volume of 3 M sodium acetate pH 5.2, followed by an equal volume of cold ethanol. Incubate for 1 h at -20 °C. Spin in a microcentrifuge at ≥ 15,000 x g for 30 min. - Resuspend DNA in 150 μl TE pH 8.0.
- Run 2 μl, 4 μl, and 8 μl of 1:10 and 1:100 dilutions on 0.8% TBE agarose gel to estimate concentration.
- Use 35 pmol of this material for the subsequent steps.
- Add 350 pmol 160Ld and 2,100 pmol 91Lg in TE, and add TE to 300 μl.
Note: The 10:1 excess of fork to template is required to prevent concatamerization, and the 6:1 excess of 91Lg to 160Ld is to ensure all ligated 160Ld is annealed to 91Lg. - Anneal fork to template by heating to 95 °C for 5 sec and let heat block cool to RT on benchtop.
- Ligate fork onto linear pUC19 by adding 15 μl T4 ligase, 36 μl 10x CutSmart buffer, 4 μl 100 mM ATP (1 mM final), adding TE to 400 μl volume. Incubate at 16 °C for at least 16 h.
- Add 10 μl 0.5 M EDTA to chelate Mg2+.
- Heat inactivate T4 ligase at 65 °C for 20 sec.
- Add 10 μl 5 M NaCl to bring [NaCl] to >100 mM (necessary to prevent nonspecific binding to Sepharose 4B column).
- Adapt syringe tip to end of column. Adapt polyethylene tubing to syringe tip and couple to fraction collector with the minimum length required for the device to reach all tubes.
- Pour a ~20 ml Sepharose 4B column and equilibrate with > 2 column volumes (CV) of cold DNA elution buffer.
- Add 420 μl reaction to 20 ml Sepharose 4B column, ensuring that buffer has completely seeped into the column bed before adding.
- After the sample has completely seeped into the column bed, elute with cold DNA elution buffer.
- Collect ~227 μl fractions (10 drops/fraction).
- Run 7 μl of each fraction on a 1% TBE agarose gel alongside a 1 kb MW marker (with gel loading buffer) at 100 V until the bromophenol blue marker reaches about 2/3 the way down.
- Stain with 2-5 μl ethidium bromide in 1x TBE and visualize on a gel imager.
Note: The gel shift of the linearized plasmid from fork tethering is very small, so do not expect to see a difference. It is also difficult to visualize free fork on this gel. We have quantified fork ligation with radiolabeled fork, and it is extremely (> 95%) efficient, thus the template should have fork attached. - Save fractions containing the ~3.2 kb band (this should be the only visible band) and measure concentration via A260 on the spectrophotometer.
- Blunt the non-fork end of the forked template with blocking oligos. Resuspend ‘blockLd’ and ‘blockLg’ to 10 μM in TE pH 8.0. Add each oligo at a 2:1 oligo: template molar ratio. Heat to 85 °C for 5 sec, cool to RT on benchtop.
Note: This step is required to prevent end-labeling of the BtsCI cut site during 32P-α-dNTP incorporation (i.e., Pol could use the non-ligated 5’ overhang as a template), however, it may be more practical to order the nucleotide-biased sequence that is removed by a blunt-cut site at this region instead. - Ligate block oligos by adding 10 μl T4 ligase. Add 1/10 volume 10x T4 ligase buffer and supplement with 1 mM ATP. Incubate at 16 °C for at least 16 h.
- Heat inactivate T4 ligase at 65 °C for 20 sec.
Primed phiX174 (model lagging strand) construction
In addition to CMG-directed leading/lagging strand synthesis, we also find it useful to use a primed, RPA-coated circular 5.4 kb ssDNA (phiX174 virion circular ssDNA) as a model lagging strand in order to isolate potential lagging strand activity in the absence of the full replisome context.- Resuspend Pr1B to 5 μM in TE pH 8.0 (see Recipes).
- Add 12 pmol Pr1B to 10 pmol phiX174 DNA (a 1.2:1 primer: template molar excess). Add buffer TE pH 8.0 to 57 μl, and 3 μl 20x SSC. Heat to 95 °C for 5 sec, cool to RT on benchtop.
- Resuspend ‘160Ld’ to 20 μM in TE buffer (see Recipes)
- Replication assays
We assume the reader has purified the full repertoire of proteins required to reconstitute the eukaryotic replisome, including at a minimum CMG helicase, Pols ε, δ and α, RFC, PCNA and RPA. Other accessory proteins such as Mcm10 and the Mrc1-Tof1-Csm3 complex will stimulate replication but are not an absolute requirement for activity. The purification and storage conditions of these proteins is beyond the scope of this protocol and can vary by specific experimental requirements, however, full purification details of the proteins are described in (Georgescu et al., 2014 and 2015) and (Langston et al., 2014). Make note of the concentrations and salt concentration of all protein stocks. Aliquot desired fractions into small (~10-25 μl) aliquots, flash freeze in liquid nitrogen, and store at -80 °C.
We prefer to use 25 μl as a standard experimental volume to minimize protein consumption while keeping the working volumes manageable. Volumes as low as 15 μl or as high as 40 μl will also work fine. For the protocols listed here, we often separate into three phases: 1) Helicase loading on the substrate. 2) Polymerase-clamp loading, i.e., polymerase, RFC clamp loader, PCNA clamp, and 2 or 3 dNTPs that allow ‘idling’ (repeated cycles of nucleotide incorporation and proofreading) of the polymerase on the template to prevent primer degradation, and 3) Replication initiation by adding the full complement of 4 dNTPs.
The first two stages may be combined if necessary, but ideally CMG loads onto forked DNA (or RPA coats ssDNA in the case of the lagging strand model) during incubation for 5-10 min or longer.
On the other hand, the second stage contains exonuclease proficient polymerases, which will degrade the substrate and/or its primer, so at least two dNTPs and a moderate incubation time with these enzymes is necessary.
The third stage initiates ongoing replication, by adding all remaining dNTPs, and in the case of fork assays, also RPA. In the examples given here, the first substrate loading phase is prepared and split into tubes as necessary, the second enzymatic phase is subsequently added, and the third initiation phase is added as the reaction timer starts. This staging method helps minimize errors derived from pipetting of small volumes, and then the splitting the mixed phases into relatively large volumes.
Because enzymes are added with respect to the final volume, it may also facilitate use of low volume such that CMG loading onto DNA can be performed at relatively high concentration. Since each phase is prepared separately before mixing, it is important to independently add salts and buffers (i.e., TDBG, MK, and any additional salts required) to each phase before adding proteins in order to prevent precipitation, aggregation, etc. that may occur if adding proteins directly to low ionic strength, depending on the protein behavior/solubility requirements.
Assuming there are differences in protein concentrations between experimental groups (e.g., during a protein titration), it is important to correct for incoming salt by adding the appropriate amount of salt to the tubes with lower protein concentrations–replication reactions are sensitive to salt conditions. Therefore, it is essential to ensure that salt has been correctly balanced.- Salt calibration
Make a standard curve of conductivity vs. salt concentration for each type of salt used in the experiment and/or protein storage, and correct experiments. - Prepare a serial dilution of salt, e.g., 0.1 M to 1 M.
- Add 10 μl each concentration to 990 μl of ddH2O.
- Measure conductivity, in mS/cm using a conductivity meter and plot against salt concentration.
- Compare conductivity of protein and/or experimental sample to standard curve. Adjust reactions with the appropriate salt as necessary.
- Leading and/or lagging strand replication; example 3-point timecourse leading/lagging (3nbf) experiment.
Note: The following protocol is identical for the 3kbf (forked linear template), apart from the addition of radionucleotides in the 3nbf experiment, as noted. Do not initiate replication until each phase and time point stop tube has been prepared. - Substrate loading (see Table 1A).
Table 1A. Substrate loading phase of leading/lagging strand replication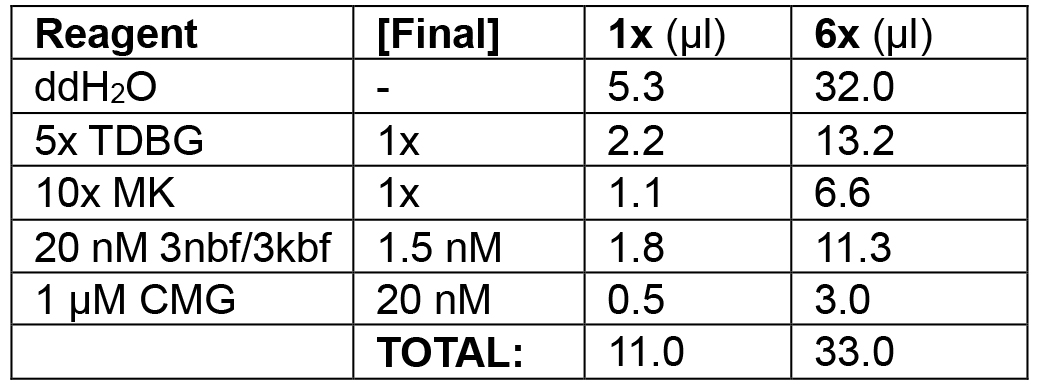
Load 0.5-2.0 nM 3nbf substrate with 10-30 nM CMG helicase (phase 1; 11 μl each).
Example: In the following order, add 32.0 μl ddH2O, 13.2 μl 5x TDBG (see Recipes) (1x final), 6.6 μl 10x MK (see Recipes) (1x final), 11.3 μl 3kbf/3nbf (1.5 nM final), and 3.0 μl 1 μM CMG (20 nM final) for a final volume of 2 groups of 3 x 11 μl = 66 μl. Incubate at 30 °C for 10 min in the water bath. Split into two groups of 33 μl.
Note: When splitting phases into separate groups, it may help to add one reaction volume to account for volume loss, e.g., split a volume that is sufficient for 7 reactions into 2 groups of 3. - Enzyme loading (see Table 1B).
Table 1B. Enzyme loading phase of leading/lagging strand replication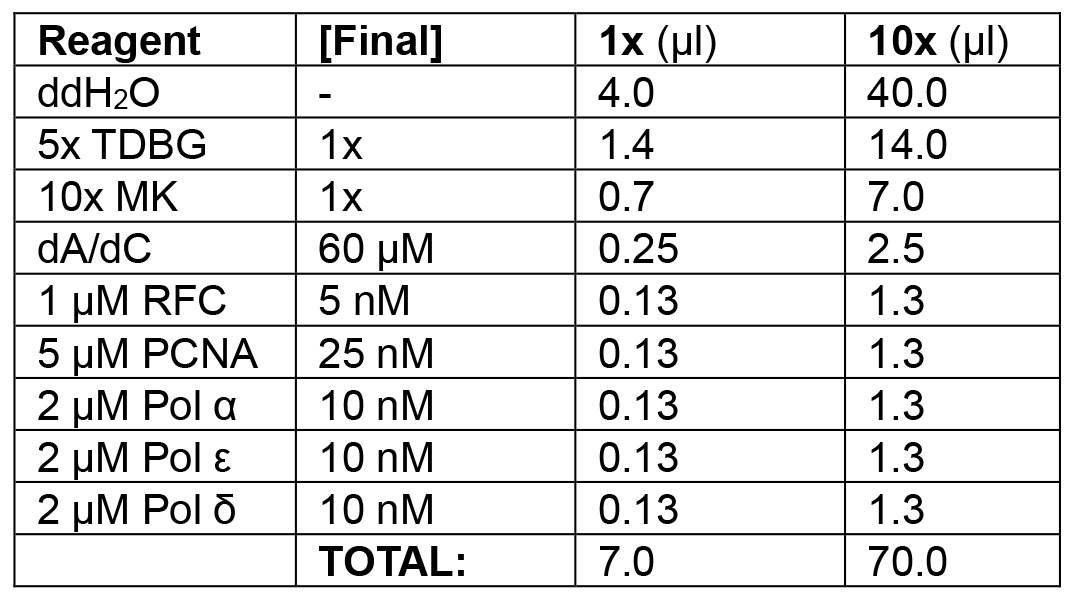
Add RFC/PCNA/Pol ε/α/δ.
Example: In the following order, add 40.0 μl ddH2O, 14.0 μl 5x TDBG (1x final), 7.0 μl 10x MK (1x final), 2.5 μl dA/dC mix (see Recipes) (60 μM final), 1.3 μl 1 μM RFC (5 nM final), 1.3 μl 5 μM PCNA (25 nM final), 1.3 μl 2 μM Pol α (10 nM final), 1.3 μl 2 μM Pol ε (10 nM final), and 1.3 μl 2 μM Pol δ (10 nM final) for a final volume of 10 x 7 μl reactions = 70 μl. Add 21 μl (3 x 7 μl) to each reaction tube, mixing thoroughly with pipette. Incubate at 30 °C for 1 min.
Note: The addition of dATP and dCTP enables polymerase idling on the primed substrate while allowing RFC to load PCNA with dATP. - Replication initiation (see Table 1C).
Table 1C. Replication initiation phase of leading/lagging strand replication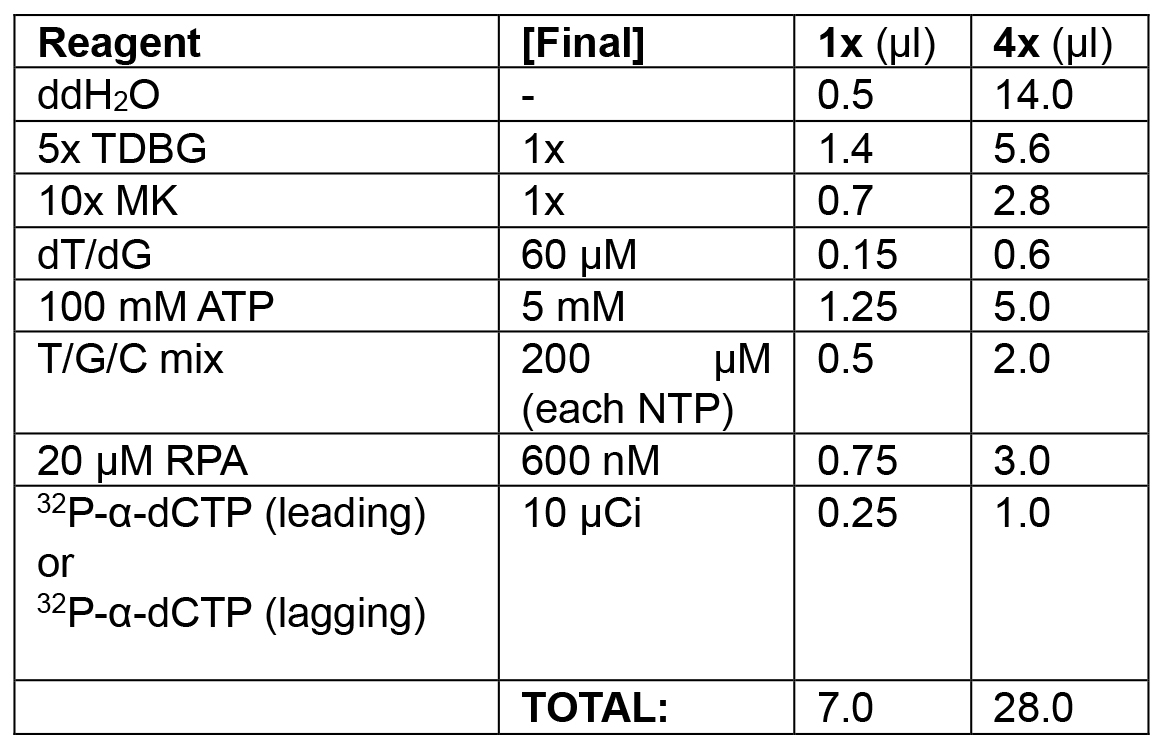
Initiate replication by adding remaining dNTPs and RPA. Monitor with 32P-α-dCTP (leading strand) or 32P-α-dGTP (lagging strand) incorporation.
Example: To each of two tubes, in the following order, add 14.0 μl ddH2O, 5.6 μl 5x TDBG (1x final), 2.8 μl 10x MK (1x final), 0.6 μl dT/dG mix (see Recipes) (60 μM final), 5 μl ATP (5 mM final), 2 μl of T/G/C mix (see Recipes) (200 μM final), and 3 μl 20 μM RPA (600 nM final). Spike the first tube (leading strand mix) with 1.0 μl 32P-α-dCTP (10 μCi/reaction). Add 1.0 μl 32P-α-dCTP to the second tube (lagging strand mix). Final volume of both initiation solutions is 28 μl, sufficient to collect time points for 4 reactions. Set a timer for 15 min. Add 21 μl of leading strand mix to the first tube; start the timer while mixing thoroughly with pipette. Add 21 μl of the lagging strand mix to the second tube. Remove 20 μl of reaction from each tube at 5, 10, and 15 min and quickly transfer to each of six tubes pre-filled with 10 μl of stop solution (see Recipes). - Model of lagging strand replication using a primed ssDNA substrate
This is a similar protocol to the above, except CMG is not included and the substrate is a primed ssDNA circle, typically phage phiX174 or M13 (or an M13 derivative). Thus, no unwinding is required (i.e., CMG is on the leading strand, not the lagging strand). The clamp, clamp loader and DNA polymerases are all stimulated by pre-coating the ssDNA with single strand binding protein, allowing one to coat the substrate with RPA beforehand, which otherwise prevents CMG loading. The primer is chosen such that a run of dC and dT is just downstream on the template, making dATP and dGTP the included nucleotides for polymerase idling.- Substrate loading (see Table 2A).
Table 2A. Substrate loading phase of lagging strand model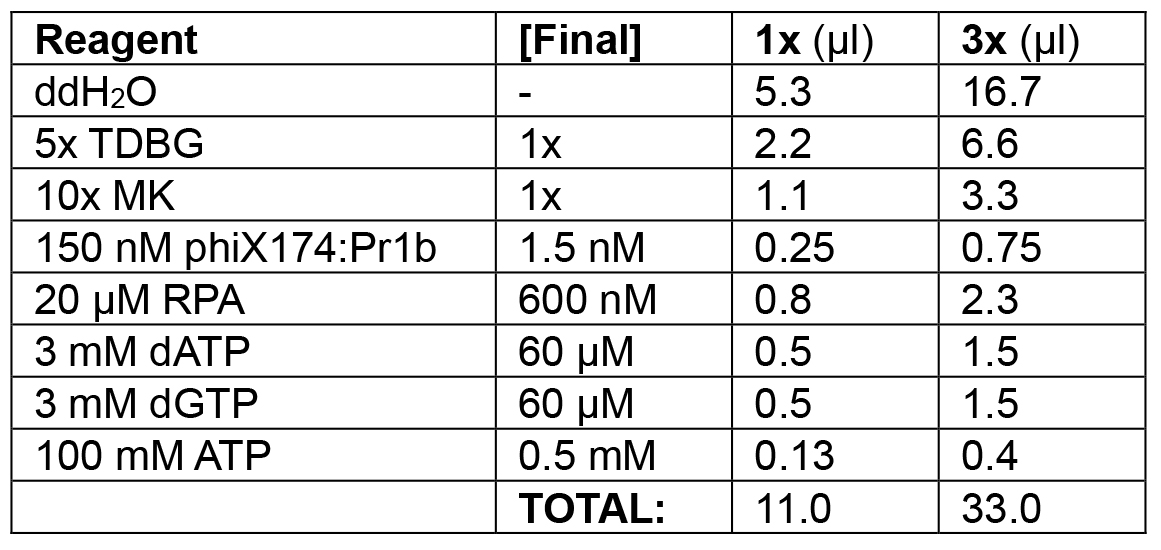
Load 0.5-2.0 nM lagging strand model substrate (phiX174:Pr1B) with 600 nM RPA (phase 1; 11 μl each).
Example: In the following order, add 16.7 μl ddH2O, 6.6 μl 5x TDBG (1x final), 3.3 μl 10x MK (1x final), 0.75 μl phiX174:Pr1b (1.5 nM final), 1.5 μl 3 mM dATP (60 μM final), 1.5 μl 3 mM dGTP (60 μM final), 0.4 μl 100 mM ATP (0.5 mM final), and 2.3 μl 20 μM RPA (600 nM final) for a final volume of 3 x 11 μl = 33 μl. Incubate at 30 °C for 10 min in the water bath. - Enzyme loading (see Table 2B).
Table 2B. Enzyme loading phase of lagging strand mode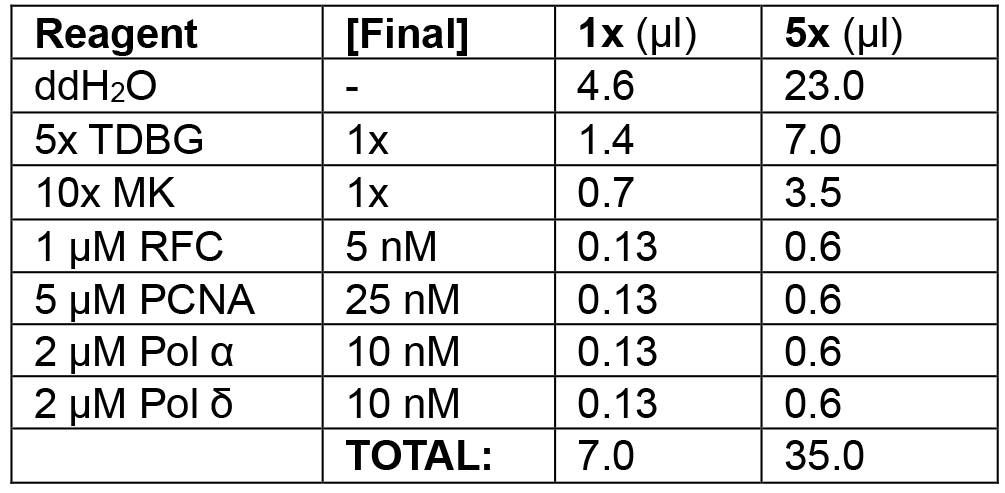
Load RFC/PCNA/α/δ.
Example: In the following order, add 23.3 μl ddH2O, 7.0 μl 5x TDBG (1x final), 3.5 μl 10x MK (1x final), 0.6 μl 1 μM RFC (5 nM final), 0.6 μl 5 μM PCNA (25 nM final), 0.6 μl 2 μM Pol α (10 nM final), and 0.6 μl 2 μM Pol δ (10 nM final) for a final volume of 5 x 7 μl reactions = 35 μl. Add 21 μl (3 x 7 μl) to each reaction tube, mixing thoroughly with a pipette tip. Incubate at 30 °C for 1 min. - Replication initiation (see Table 2C).
Table 2C. Replication initiation phase of lagging strand model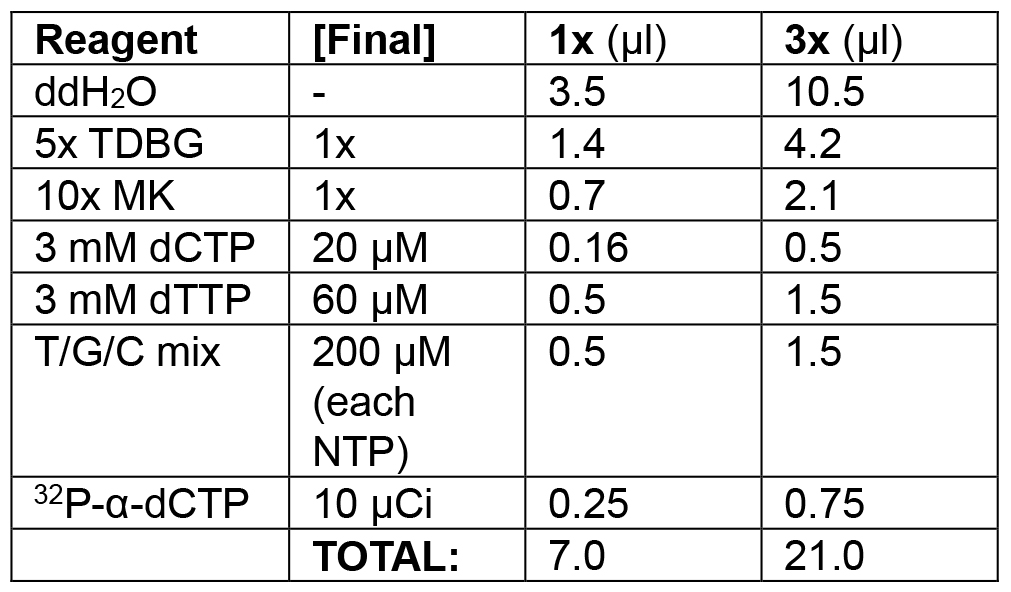
Initiate replication by adding the nucleotides that had been withheld during preincubation.
Example: in the following order, add 10.5 μl ddH2O, 4.2 μl 5x TDBG (1x final), 2.1 μl 10x MK (1x final), 0.5 μl 3 mM dCTP (20 μM final), 1.5 μl 3 mM dTTP (60 μM final), 1.5 μl T/C/G mix (200 μM final), 2 μl of T/G/C mix (200 μM final). Spike with 0.75 μl 32P-α-dCTP (10 μCi/reaction). Set a timer for 3 min. Add 21 μl of initiation mix; start timer while mixing thoroughly with pipette. Remove 20 μl of reaction at 1, 2, and 3 min and quickly transfer to each of three tubes pre-filled with 10 μl of stop solution.
- Substrate loading (see Table 2A).
- Replication product visualization
For 3nbf experiments, leading strand products should look similar to those in Figure 1b of Georgescu et al. (2015); i.e., products whose length increases over time and goes to full length (3.2 kb) by ~10 min or less. Using an end-labeled primer to monitor primer extension on the 3kbf substrate, results should look similar to those in Figure 3b of Schauer et al. (2017); i.e., products whose length increases over time and goes to full length (2.8 kb). Replisome-directed lagging strand replication should appear similar to those in Figure 6 of Schauer et al. (2017); i.e., ~200 bp products. For all CMG-directed experiments, subtraction of either CMG or ATP from the experiment is a good negative control and should yield no extended product. Using the lagging strand model substrate, the experiments should look similar to those in Figure 1b in Schauer et al. (2017); i.e., robust full length (5.4 kb) product in under 3 min.- Prepare ladder
Follow manufacturer’s protocols to 5’-end-label the 1 kb DNA ladder with T4 kinase and 32P-γ-ATP. Typically, we label 20 μl of the ladder in 50 μl total with 5 μl T4 PNK and 4 μl 32P-γ-ATP. Purify away free 32P-γ-ATP with a G-25 column that has been equilibrated with TE, according to the manufacturer’s protocol. - Prepare alkaline agarose gel
Add 2.6 or 1.6 g LE agarose to 200 ml distilled H2O (1.3% for 3nbf/3kbf substrates, 0.8% for lagging strand model substrates). Microwave until boiling. Cool to at least 60 °C and add 0.5 ml 0.5 M EDTA and 0.6 ml 10 N NaOH before pouring the gel. Let cool to room temperature (RT) over ~2 h. Submerge in alkaline running buffer (see Recipes) in horizontal gel box. - Load samples and DNA standard in gel. Run for 16 h at 35 V.
- Remove free radionucleotides
Remove gel from gel box and expose to phosphorimaging screen for 15 min. Scan on phosphorimager and print gel at full size. Cut the saturated portion off of the printout, overlay it on the gel, and cut the bottom part of the gel off that contains free radionucleotides (usually the bottom third or fourth) in order to reduce exposure of yourself and your lab equipment. The identification of this region is particularly important when monitoring lagging strand synthesis as free radionucleotides do not run too far below Okazaki fragments. Free radionucleotides can also be run off the gel, but it contaminates the equipment, creates further radioactive waste, and they tend to diffuse in the buffer and can leach back into the entire gel and contribute to background signal. - Compress the gel
Wrap plastic wrap tightly around a flat surface like the bottom of a plexiglass radiation shield. Place the gel slab on the saran wrap. Place C-fold paper towels flat; one adjacent to each gel edge. Place one sheet of DEAE-cellulose paper over gel. Place one or two sheets of Whatman 3MM chromatography transfer paper over this. Stack 2 horizontal rows of ~25 C-fold paper towels over this. Cover paper towels with something flat and rigid (e.g., a flat plexiglass shield) to distribute weight. Gently rest a heavy weight (e.g., a lead brick) on the center of this assembly and press thin (few mm thick) for 6-12 h. We find this procedure superior to using a gel dryer, and it doubles as a step to allow DNA to transfer and immobilize on the DEAE-cellulose paper. - Remove all paper except DEAE-cellulose, wrap in plastic wrap, and expose gel face to phosphorimaging screen for 2-36 h, depending on isotope strength.
- Scan phosphorimaging screen on a phosphorimager.
- Prepare ladder
- Salt calibration
Data analysis
Analysis of primer extension experiments is straightforward and well-documented elsewhere, and most operations can be handled by the Gels package in ImageJ (see https://imagej.nih.gov/ij/docs/menus/analyze.html#gels). Parameters of interest may be velocity, primer uptake, product length, etc., and will depend on the experiment. In addition to reading the documentation of the Gels package, we strongly encourage the reader to read through the ImageJ User Guide (https://imagej.nih.gov/ij/docs/guide/) for general help with image analysis before analyzing gel data.
When using radionucleotide incorporation, it is crucial to correct the bias of 32P incorporation using the DNA ladder as a reference point, as longer products will have much more incorporated nucleotide signal on a molar basis of short relative to long DNA molecules (Kurth et al., 2013 and Georgescu et al., 2014). The consequence of not performing this step is thus an overestimation of longer replication products and an underestimation of shorter ones. Use the following routine to quantify the gels, as in as in Figure 1c of Georgescu et al. (2015).
- Extract the lane intensity profiles (i.e., average pixel intensity vs. vertical distance) from the gel scans using the ImageJ.
- Rotate the gel horizontally so that lanes run left to right. Image>Transform>Rotate 90° right.
- Select the rectangle icon on the ImageJ toolbar and make a rectangle around the first lane.
- Plot the integrated intensity profile (average intensity in a.u. vs. distance in cm). Analyze>Plot Profile (or Ctrl+K).
- A new window will appear. Save profile as a text file by clicking save.
- Move rectangle down to next lane with the down arrow and repeat step 1c. Keeping an identical rectangle size and horizontal position assures the gel lanes will line up correctly.
- Import the intensity profiles as tab delimited data into spreadsheet software.
- Rotate the gel horizontally so that lanes run left to right. Image>Transform>Rotate 90° right.
- Convert the distance in the intensity profiles to DNA lengths.
- Fit the lane with the molecular weight (MW) standard with a probability distribution containing the same number of Gaussian peaks as the number of bands that appear in the DNA MW standard.
- Plot the length of the dsDNA standards (bp) vs. the mean vertical distance (cm) obtained from the Gaussian fits, and fit the data with a logarithmic function, making note of the equation of the fitted line.
- In a spreadsheet, convert vertical distance (cm) to DNA (bp) using the equation obtained in Data analysis step 1c.
- Fit the lane with the molecular weight (MW) standard with a probability distribution containing the same number of Gaussian peaks as the number of bands that appear in the DNA MW standard.
- Correct the lane intensity profiles for molecular weight. Normalize the intensity of the products on a pixel-by-pixel basis by dividing each intensity value by its DNA length (as determined in step 2) and multiplying by the full template length (in bp). The resulting profiles can be curve fit with any software capable of nonlinear curve fitting, and analyzed as necessary on a case-by-case basis.
Notes
- In preparing the 3nbf substrate, it is critical to remove all potential sources of end-labeling, including nicks in the template and flaps due to inefficient fork ligation. We therefore recommend extremely gentle handling of the template including minimal vortexing, gentle pipetting, etc.
- Extended ligations during the fork ligation step are useful. End labeling presents as a full-length band that appears at early timepoints of replication assays; this should be absent or only marginally detectable if the substrate has been correctly prepared. As a control, perform reactions lacking CMG and/or ATP to assess the degree of end-labeling; without CMG unwinding activity there should be no appearance of full-length product at any time, since Pol ε cannot strand displace.
- Nucleases pose a major problem for replication assays as they degrade the substrates and also contribute to end labeling, which can be indistinguishable from full length primer extension if the signal intensity is too high. It is therefore critical to check all protein preps and reagents for nucleases. Microbial contamination is another major culprit for bringing in nucleases, so ensure that all reagents are autoclaved, filter sterilized, aliquotted for single use, and frozen. There are commercial kits available to test DNAse and RNAse activity, however, a timecourse of incubation of your reagent of interest with a 32P-labeled oligo (such as the primer created for 3kbf) will also be informative. Nuclease activity will appear as a time-dependent disappearance of 32P signal on a nondenaturing polyacrylamide gel.
Recipes
Note: Store all solutions at -20 °C unless otherwise specified. Buffers used in replication assays should not be stored longer than ~6 weeks.
- 6x gel loading dye
60% glycerol
0.1% bromophenol blue
0.1% xylene cylanol
Store at RT - TE buffer, pH 8.0
10 mM Tris-HCl, pH 8.0
1 mM EDTA (Ethylenediaminetetraacetic acid, disodium salt) - 10x TBE
1 M Tris base
1 M boric acid
20 mM EDTA - DNA elution buffer
100 mM NaCl
10 mM Tris-HCl, pH 8.0
1 mM EDTA - 20x SSC
3 M NaCl
0.3 M sodium citrate - 1-Butanol saturated water
50 ml 1-butanol
5 ml ddH2O
Shake vigorously and store at 4 °C - 5x TDBG
125 mM Tris-acetate, pH 7.5
25% glycerol
200 μg/ml BSA
10 mM TCEP (Tris(2-carboxyethyl)phosphine) pH 7.5
15 mM DTT
0.5 mM EDTA (Ethylenediaminetetraacetic acid) - 10x MK
500 mM potassium glutamate
100 mM magnesium acetate - dA/dC mix
10 mM dATP
10 mM dCTP
10 mM Tris-HCl, pH 7.5
1 mM EDTA - dT/dG mix
10 mM dTTP
10 mM dGTP
10 mM Tris-HCl, pH 7.5
1 mM EDTA - T/G/C mix
10 mM TTP
10 mM GTP
10 mM CTP
10 mM Tris-HCl, pH 7.5
1 mM EDTA - Stop solution
1% SDS
40 mM EDTA
10% glycerol
0.02% bromophenol blue
0.02% xylene cylanol
Store at RT - Alkaline running buffer
1.125 mM EDTA
30 mM NaOH
Acknowledgments
This work was supported by grants from the National Institutes of Health (T32 CA009673 to G.S; and R01 GM115809 and the Howard Hughes Medical Institute to M.O.D.). The protocols herein were adapted from those originally used in Georgescu et al., 2014, Georgescu et al., 2015, and Schauer et al., 2017.
References
- Georgescu, R. E., Langston, L., Yao, N. Y., Yurieva, O., Zhang, D., Finkelstein, J., Agarwal, T. and O’Donnell, M. E. (2014). Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat Struct Mol Biol 21(8): 664-670.
- Georgescu, R. E., Schauer, G. D., Yao, N. Y., Langston, L. D., Yurieva, O., Zhang, D., Finkelstein, J. and O’Donnell, M. E. (2015). Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. Elife 4: e04988.
- Kurth, I., Georgescu, R. E. and O'Donnell, M. E. (2013). A solution to release twisted DNA during chromosome replication by coupled DNA polymerases. Nature 496(7443): 119-122.
- Langston, L. D. and O’Donnell, M. (2008). DNA polymerase δ is highly processive with proliferating cell nuclear antigen and undergoes collision release upon completing DNA. J Biol Chem 283(43): 29522-29531.
- Langston, L. D., Zhang, D., Yurieva, O., Georgescu, R. E., Finkelstein, J., Yao, N. Y., Indiani, C. and O’Donnell, M. E. (2014). CMG helicase and DNA polymerase ε form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc Natl Acad Sci U S A 111(43): 15390-15395.
- Schauer, G. D. and O'Donnell, M. E. (2017). Quality control mechanisms exclude incorrect polymerases from the eukaryotic replication fork. Proc Natl Acad Sci U S A 114(4): 675-680.
- Stodola, J. L. and Burgers, P. M. (2016). Resolving individual steps of Okazaki-fragment maturation at a millisecond timescale. Nat Struct Mol Biol 23(5): 402-408.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schauer, G., Finkelstein, J. and O’Donnell, M. (2017). In vitro Assays for Eukaryotic Leading/Lagging Strand DNA Replication. Bio-protocol 7(18): e2548. DOI: 10.21769/BioProtoc.2548.
Category
Biochemistry > Protein > Activity
Molecular Biology > DNA > DNA synthesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.