- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Method to Convert mRNA into a Guide RNA (gRNA) Library without Requiring Previous Bioinformatics Knowledge of the Organism
.jpg)
Published: Vol 7, Iss 10, May 20, 2017 DOI: 10.21769/BioProtoc.2319 Views: 10768
Reviewed by: Jihyun KimManuela RoggianiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
While the diversity of species represents a diversity of special biological abilities, many of the genes that encode those special abilities in a variety of species are untouched, leaving an untapped gold mine of genetic information; however, despite current advances in genome bioinformatics, annotation of that genetic information is incomplete in most species, except for well-established model organisms, such as human, mouse, or yeast. A guide RNA (gRNA) library using the clustered regularly interspersed palindromic repeats (CRISPR)/Cas9 (CRISPR-associated protein 9) system can be used for the phenotypic screening of uncharacterized genes by forward genetics. The construction of a gRNA library usually requires an abundance of chemically synthesized oligos designed from annotated genes; if one wants to convert mRNA into gRNA without prior knowledge of the target DNA sequences, the major challenges are finding the sequences flanking the protospacer adjacent motif (PAM) and cutting out the 20-bp fragment. Recently, I developed a molecular biology-based technique to convert mRNA into a gRNA library (Arakawa, 2016) (Figure 1). Here I describe the detailed protocol of how to construct a gRNA library from mRNA.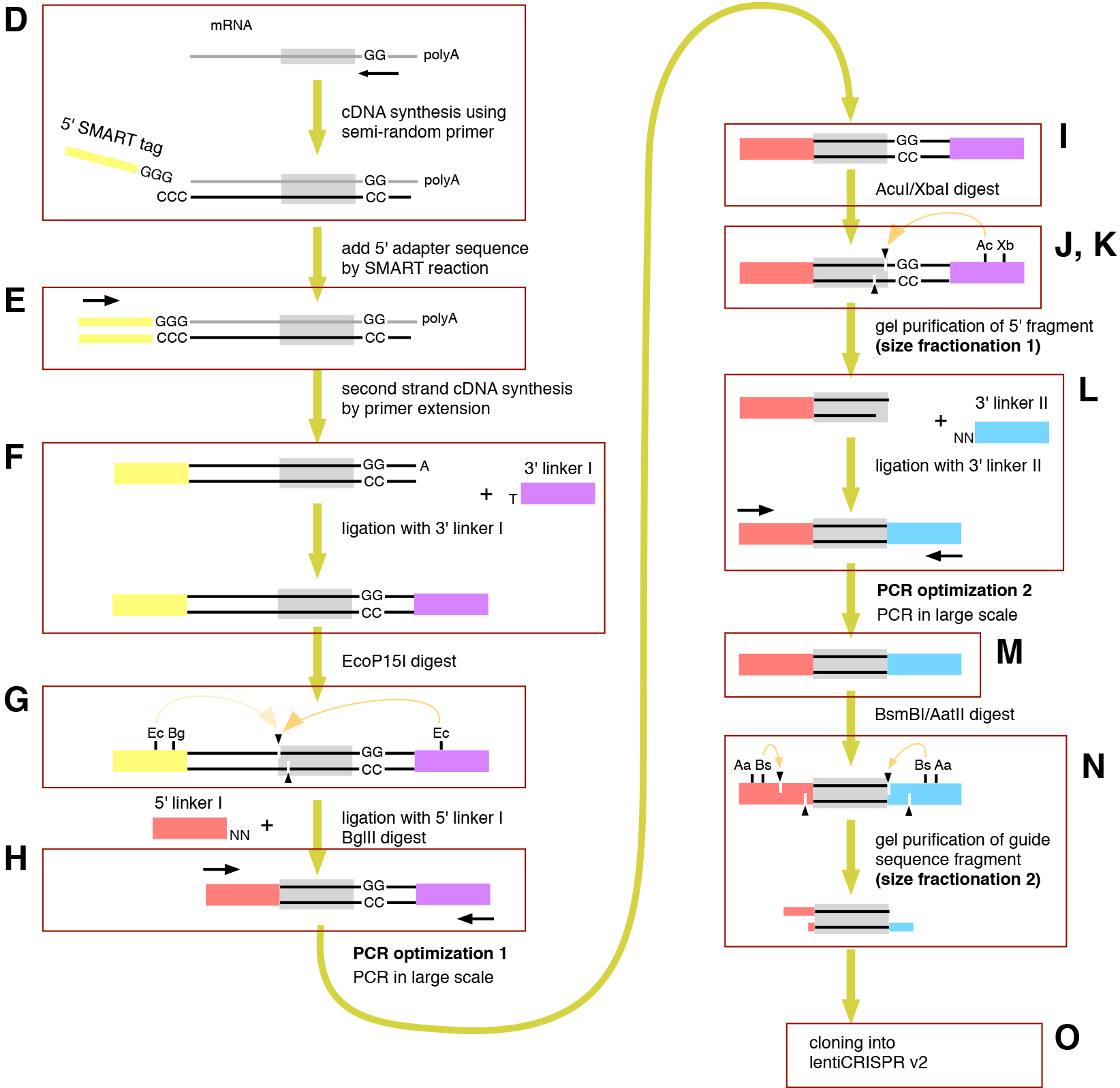
Figure 1. A method to convert mRNA into a gRNA library construction (Sanjana et al., 2014). The scheme of the method is summarized. Each step of D-O is described in detail in the Procedure. Bg, BglII; Xb, XbaI; Bs, BsmBI; Aa, AatII. PCR, polymerase chain reaction; lentiCRISPR v2, lentiCRISPR version 2.
Background
The clustered regularly interspersed palindromic repeats (CRISPR) system is responsible for the acquired immunity of bacteria (Barrangou et al., 2007), which is shared among 40% of eubacteria and 90% of archaea (Grissa et al., 2007). While CRISPR/Cas9 is, physiologically, an endonuclease used to eliminate the infectious pathogen (Barrangou et al., 2007), CRISPR/Cas9 can be used to cleave any locus of the genome if a guide RNA (gRNA) is provided (Cong et al., 2013; Mali et al., 2013). By designing gRNA for the gene of interest, individual genes can be knocked out one-by-one by non-homologous end joining (NHEJ) (Cong et al., 2013; Mali et al., 2013); additionally, CRISPR/Cas9 can be utilized to make a gRNA library available for genetic screening (Zhou et al., 2001; Koike-Yusa et al., 2014; Shalem et al., 2014; Wang et al., 2014). The gRNA for Streptococcus pyogenes (Sp) Cas9 can be designed as a 20-bp sequence adjacent to the protospacer adjacent motif (PAM) NGG (Cong et al., 2013; Mali et al., 2013). Such a sequence can usually be identified from the coding sequence or locus of interest by bioinformatics techniques. Here, I describe a method to construct a gRNA library via molecular biology techniques without relying on bioinformatics. Briefly, one synthesizes cDNA from the extracted RNA using a semi-random primer containing a PAM-complementary sequence and then cuts out the 20-mer adjacent to the PAM using type IIS and type III restriction enzymes to create a gRNA library. The described approach does not require prior knowledge about the target DNA sequences, making it applicable to any species.
Materials and Reagents
- 1.5 ml microcentrifuge tube
- 0.2 ml PCR tube
- Disposable pipette tip
- OligodT column (QIAGEN, supplemented with the Oligotex mRNA Mini Kit [QIAGEN, catalog number: 70022 ])
- STBL4 electro-competent cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11635018 )
- Lentiviral vector
lentiCRISPR v2 (Sanjana et al., 2014) (Addgene, catalog number: 52961 ) - Oligonucleotides
Semi-random primer p NNNCCN
5’ switching mechanism at RNA transcript (SMART) tagTGGTCAAGCTTCAGCAGATCTACACGGACGTCGCrGrGrG
5’ SMART PCR primer TGGTCAAGCTTCAGCAGATCTACACG
3’ linker I forward p CTGCTGACTTCAGTGGTTCTAGAGGTGTCCAA
3’ linker I reverse GTTGGACACCTCTAGAACCACTGAAGTCAGCAGT
5’ linker I forward GCATATAAGCTTGACGTCTCTCACCG
5’ linker I reverse p NNCGGTGAGAGACGTCAAGCTTATATGC
3’ linker II forward p GTTTGGAGACGTCTTCTAGATCAGCG
3’ linker II reverse CGCTGATCTAGAAGACGTCTCCAAACNN
3’ linker I PCR primer GTTGGACACCTCTAGAACCACTGAAGTCAGCAGTNNNCC
3’ linker II PCR primer CGCTGATCTAGAAGACGTCTCCAAAC
LentiCRISPR forward CTTGGCTTTATATATCTTGTGGAAAGGACG
LentiCRISPR reverse CGGACTAGCCTTATTTTAACTTGCTATTTCTAG - TRIzol reagent (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15596026 )
- Phenol:chloroform:isoamyl alcohol 25:24:1 (Sigma-Aldrich, catalog number: P2069-100ML )
- Ethanol
- RNase-free water
- Oligotex mRNA Mini Kit (QIAGEN, catalog number: 70022 )
- T4 DNA ligase reaction buffer (New England Biolabs, catalog number: B0202S )
- SMART Scribe reverse transcriptase (Takara Bio, Clontech, catalog number: 639536 )
- DTT (Takara Bio, supplemented with SMART Scribe reverse transcriptase [Takara Bio, Clontech, catalog number: 639536 ])
- dNTP mix (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18427013 )
- RNaseOUT (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10777019 )
- RNase H (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18021014 )
- MilliQ water
- Advantage 2 polymerase mix (Takara Bio, Clontech, catalog number: 639201 )
- QIAquick PCR Purification Kit (QIAGEN, catalog number: 28104 )
- Quick Ligation Kit (New England Biolabs, catalog number: M2200S )
- EcoP15I (New England Biolabs, catalog number: R0646S )
- BglII (New England Biolabs, catalog number: R0144S )
- AcuI (New England Biolabs, catalog number: R0641S )
- XbaI (New England Biolabs, catalog number: R0145S )
- BsmBI (New England Biolabs, catalog number: R0580S )
- AatII (New England Biolabs, catalog number: R0117S )
- 10-bp ladder
- 1x CutSmart buffer (included in XbaI [New England Biolabs, catalog number: R0145S ])
- S-adenosylmethionine (SAM) (New England Biolabs, supplemented with AcuI [New England Biolabs])
- 3 M sodium acetate (pH 5.5) (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9740 )
- Acrylamide/Bis solution (19:1) (40 % w/v, 5 % C) (SERVA Electrophoresis, catalog number: 10679.01 )
- Glycogen (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0561 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32851 )
- TE buffer (pH 8.0) (see Recipes)
Equipment
- Pipettes
- Centrifuge (Eppendorf, models: 5424 R , 5810 R )
- Heating block
- Glass beaker
- GeneAmp PCR System 9700 (Thermo Fisher Scientific, model: GeneAmp PCR System 9700 )
Note: This product has been discontinued. - Mini-PROTEAN Tetra Vertical Electrophoresis Cell (Bio-Rad Laboratories, model: Mini-PROTEAN® Tetra Vertical Electrophoresis Cell )
- GenePulser II (Bio-Rad Laboratories, model: Gene Pulser II )
- High Performance Laboratory Incubator–Mod. 2800 (F.lli GALLI, model: MOD. 2800 )
- CLC Genomics Workbench (QIAGEN, model: CLC Genomics Workbench )
- Bioanalyzer (Agilent Technologies)
- Autoclave
Procedure
- Total RNA preparation
- Lyse 108 cells with 10 ml of TRIzol by repetitive pipetting. Incubate for 5 min at room temperature.
- Add 2 ml of chloroform. Shake by hand for 15 sec. Incubate for 2-3 min at room temperature. Centrifuge at 12,000 x g for 15 min at 4 °C.
- Transfer the aqueous phase to a fresh tube. Add 5 ml of isopropyl alcohol and mix well. Centrifuge at 12,000 x g for 10 min at 4 °C.
- Remove the supernatant. Wash the pellet once with 80% ethanol. Air dry the pellet for 5-10 min.
- Dissolve RNA in 1 ml of RNase-free water by pipetting, then incubate the sample for 10 min at 55-60 °C.
- Lyse 108 cells with 10 ml of TRIzol by repetitive pipetting. Incubate for 5 min at room temperature.
- PolyA RNA preparation (see Note 1)
- Use the Oligotex mRNA Midi Kit. Heat the Oligotex suspension to 37 °C. Heat buffer OEB to 70 °C on a heating block. Mix buffer OBB well.
- Pipet 500 µg of total RNA into an RNase-free 1.5-ml microcentrifuge tube and adjust the volume with RNase-free water to 1 ml.
- Add 500 µl of buffer OBB and 30 µl of the Oligotex suspension.
- Incubate for 3 min at 70 °C. Remove from heat.
- Incubate for 10 min at room temperature.
- Centrifuge for 2 min at 15,871 x g. Remove supernatant by pipetting.
- Add 250 µl of RNase-free water and 250 µl of buffer OBB. Mix the contents thoroughly by pipetting.
- Incubate for 3 min at 70 °C. Remove from heat.
- Incubate for 10 min at room temperature.
- Centrifuge for 2 min at 15,871 x g. Remove supernatant by pipetting.
- Resuspend the pellet in 400 µl of buffer OW2. Pipet the mixture onto a small spin column placed in a 1.5-ml tube. Centrifuge for 1 min at 15,871 x g.
- Transfer the spin column to a new RNase-free 1.5-ml tube. Apply 400 µl of buffer OW2. Pipet onto a small spin column placed in a 1.5-ml tube. Centrifuge for 1 min at 15,871 x g.
- Transfer the spin column to a new RNase-free 1.5-ml tube. Pipet 25 µl of hot (70 °C) buffer OEB onto the column. Pipet up and down 3-4 times to resuspend the resin. Centrifuge for 1 min at 15,871 x g.
- Pipet another 25 µl of hot (70 °C) buffer OEB onto the column. Pipet up and down 3-4 times to resuspend the resin. Centrifuge for 1 min at 15,871 x g.
- Use the Oligotex mRNA Midi Kit. Heat the Oligotex suspension to 37 °C. Heat buffer OEB to 70 °C on a heating block. Mix buffer OBB well.
- Linker preparation
- Combine the following reagents in a 1.5-ml microcentrifuge tube: 10 µl of 100 µM linker forward oligo, 10 µl of 100 µM linker reverse oligo, and 2.2 µl of 10x T4 DNA ligase buffer (New England Biolabs).
- Place the tubes in a glass beaker containing 2 L of boiled water and incubate the tubes until the water cools down to room temperature naturally.
- Dilute the annealed oligos with 77.8 µl of TE buffer (pH 8.0) and use as 10 µM linkers.
- Combine the following reagents in a 1.5-ml microcentrifuge tube: 10 µl of 100 µM linker forward oligo, 10 µl of 100 µM linker reverse oligo, and 2.2 µl of 10x T4 DNA ligase buffer (New England Biolabs).
- First-strand cDNA synthesis
- Combine the following reagents in a 0.2 ml PCR tube: 200 ng of poly(A) RNA (from step A2), 0.6 µl of 25 µM semi-random primers, and RNase-free water in a 4.75 µl volume.
- Incubate the tube at 72 °C in a hot-lid thermal cycler for 3 min, cool on ice for 2 min, and further incubate at 25 °C for 10 min.
- Increase the temperature to 42 °C and add a 5.25 µl mixture of the following reagents: 0.5 µl of 25 µM 5’ SMART tag, 2 µl of 5x SMART Scribe buffer, 0.25 µl of 100 mM DTT, 1 µl of 10 mM dNTP mix, 0.5 µl of RNaseOUT (Invitrogen), and 1 µl SMART Scribe reverse transcriptase (100 U) (Clontech).
- Incubate the first-strand cDNA reaction mixture at 42 °C for 90 min and then at 68 °C for 10 min. To degrade RNA, add 1 µl of RNase H (Invitrogen) to the mixture and incubate the mixture at 37 °C for 20 min.
- Combine the following reagents in a 0.2 ml PCR tube: 200 ng of poly(A) RNA (from step A2), 0.6 µl of 25 µM semi-random primers, and RNase-free water in a 4.75 µl volume.
- Double-stranded (ds) cDNA synthesis by primer extension
- Mix 11 µl of the prepared first-strand poly(A) cDNA (from step C4) with 74 µl of MilliQ water, 10 µl of 10x Advantage 2 PCR buffer, 2 µl of 10 mM dNTP mix, 1 µl of 25 µM 5’ SMART PCR primer, and 2 µl of 50x Advantage 2 polymerase mix (Clontech).
- Incubate a 100 µl volume of the reaction mixture for primer extension at 95 °C for 1 min, 68 °C for 20 min, and then 70 °C for 10 min.
- Purify the prepared ds cDNA using a QIAquick PCR Purification Kit (QIAGEN) and elute the DNA with 40 µl of TE buffer (pH 8.0).
- Mix 11 µl of the prepared first-strand poly(A) cDNA (from step C4) with 74 µl of MilliQ water, 10 µl of 10x Advantage 2 PCR buffer, 2 µl of 10 mM dNTP mix, 1 µl of 25 µM 5’ SMART PCR primer, and 2 µl of 50x Advantage 2 polymerase mix (Clontech).
- 3’ linker I ligation
- Mix ds poly(A) cDNA (from step D3) with 0.5 µl of 10 µM 3’ linker I and 1 µl of Quick T4 DNA ligase (New England Biolabs; NEB) in 1x Quick ligation buffer.
- Incubate the ligation reaction mixture at room temperature for 15 min, then purify it using a QIAquick PCR Purification Kit and elute it with 80 µl of TE buffer.
- Mix ds poly(A) cDNA (from step D3) with 0.5 µl of 10 µM 3’ linker I and 1 µl of Quick T4 DNA ligase (New England Biolabs; NEB) in 1x Quick ligation buffer.
- EcoP15I digestion
- Digest the 3’ linker I-ligated DNA (from step E2) with 1 µl EcoP15I (10 U/µl, NEB) in 1x NEBuffer 3.1 containing 1x ATP in a 100 µl volume at 37 °C overnight.
- Purify the EcoP15I-digested DNA using a QIAquick PCR Purification Kit and elute the DNA with 40 µl of TE buffer.
- Digest the 3’ linker I-ligated DNA (from step E2) with 1 µl EcoP15I (10 U/µl, NEB) in 1x NEBuffer 3.1 containing 1x ATP in a 100 µl volume at 37 °C overnight.
- 5’ linker I ligation and BglII digestion
- Mix the digested DNA (from step F2) with 0.5 µl of 10 µM 5’ linker I and 1 µl of Quick T4 DNA ligase (NEB) in 1x Quick ligation buffer.
- Incubate the ligation reaction mixture at room temperature for 15 min, purify it using a QIAquick PCR Purification Kit, and elute it with 80 µl of TE buffer.
- Digest the DNA with 1 µl of BglII (10 U/µl, NEB) (see Note 2) in 1x NEBuffer 3.1 in a 100 µl volume at 37 °C for 3 h.
- Purify the EcoP15I/BglII-digested DNA using a QIAquick PCR Purification Kit and elute the DNA with 50 µl of TE buffer.
- Mix the digested DNA (from step F2) with 0.5 µl of 10 µM 5’ linker I and 1 µl of Quick T4 DNA ligase (NEB) in 1x Quick ligation buffer.
- First PCR optimization (see Note 3)
- To determine the optimal number of PCR cycles, prepare a 0.2 ml PCR tube containing 5 µl of the ds cDNA ligated with 5’ linker I/3’ linker I (from step G4), 0.5 µl of 25 µM 5’ linker I forward primer, 0.5 µl of 25 µM 3’ linker I PCR primer, 5 µl of 1x Advantage 2 PCR buffer, 1 µl of 10 mM dNTP mix, 1 µl of 50x Advantage 2 polymerase mix, and MilliQ water in a 50 µl volume.
- Perform PCR with the following cycling parameters: 6 cycles of 98 °C for 10 sec and 68 °C for 10 sec.
- After the 6 cycles, transfer 5 μl of the reaction to a clean microcentrifuge tube. Perform PCR with the rest of the PCR reaction mixture for 3 additional cycles of 98 °C for 10 sec and 68 °C for 10 sec.
- After these additional 3 cycles, transfer 5 μl to a clean microcentrifuge tube.
- In the same way, repeat additional PCR until reaching 30 total cycles. Thus, prepare a series of PCR reactions of 6, 9, 12, 15, 18, 21, 24, 27, and 30 cycles and analyze by 20% polyacrylamide gel electrophoresis to compare the band patterns (Figure 2; PCR optimization 1).
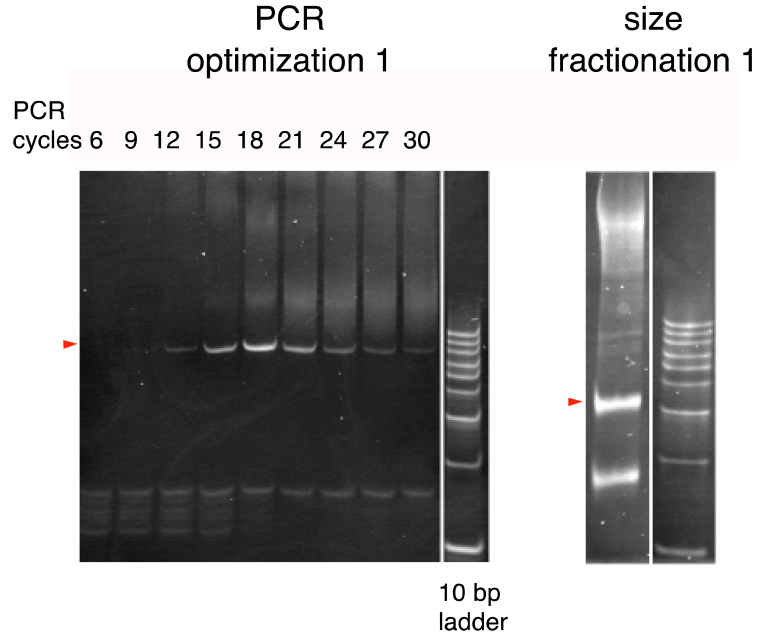
Figure 2. The first PCR cycle optimization and size fractionation (Arakawa, 2016). PCR products were run on 20% polyacrylamide gels. A 10-bp ladder was used as the size marker. Bands of the expected sizes are marked by triangles. (see Note 4) - Determine the optimal number of PCR cycles as the minimal number of PCR cycles yielding the greatest quantity of the 84-bp product (typically around 17 cycles).
- Repeat two 50-µl PCR reactions with the optimal number of PCR cycles.
- Purify the PCR product using a QIAquick PCR Purification Kit and elute with 50 µl of TE buffer.
- To determine the optimal number of PCR cycles, prepare a 0.2 ml PCR tube containing 5 µl of the ds cDNA ligated with 5’ linker I/3’ linker I (from step G4), 0.5 µl of 25 µM 5’ linker I forward primer, 0.5 µl of 25 µM 3’ linker I PCR primer, 5 µl of 1x Advantage 2 PCR buffer, 1 µl of 10 mM dNTP mix, 1 µl of 50x Advantage 2 polymerase mix, and MilliQ water in a 50 µl volume.
- AcuI/XbaI digestion
- Digest the PCR product (from step H8) with 2 µl of AcuI (5 U/µl, NEB) and 2 µl of XbaI (20 U/µl, NEB) (see Note 2) in 1x CutSmart buffer containing 40 µM S-adenosylmethionine (SAM) in a 60 µl volume at 37 °C overnight.
- Digest the PCR product (from step H8) with 2 µl of AcuI (5 U/µl, NEB) and 2 µl of XbaI (20 U/µl, NEB) (see Note 2) in 1x CutSmart buffer containing 40 µM S-adenosylmethionine (SAM) in a 60 µl volume at 37 °C overnight.
- The crush and soak procedure
- Run the AcuI/XbaI-digested DNA (from step I1) on a 20% polyacrylamide gel (Figure 2; size fractionation 1).
- Cut the 45-bp fragment out of the gel and transfer it to a microfuge tube. Crush the gel slice against the wall of the microfuge tube with the disposable pipette tip.
- Add 1-2 volumes of TE to the gel slice. Incubate the tube at room temperature overnight on a rotating wheel.
- Centrifuge the sample at 21,130 x g for 1 min at 4 °C. Transfer the supernatant to a fresh microfuge tube, being careful to avoid transferring fragments of polyacrylamide.
- Add 0.5 volumes of TE to the polyacrylamide pellet. Vortex the tube briefly. Centrifuge the sample at 21,130 x g for 1 min at 4 °C.
- Combine the two supernatants and then centrifuge at 21,130 x g for 1 min at 4 °C. Transfer the supernatant to the new microcentrifuge tube in order to remove the small PAGE debris.
- Mix the sample with 0.1 volume of 3 M sodium acetate and 1/400 volume of 20 µg/µl of glycogen. Add 2.5 volumes of cold ethanol to the sample and mix briefly. Store the solution on ice for 30 min. Centrifuge the sample at 21,130 x g for 20 min at 4 °C.
- Discard the supernatant. Rinse the pellet once with 70% ethanol.
- Dissolve the pellet into 20 µl of TE buffer.
- Run the AcuI/XbaI-digested DNA (from step I1) on a 20% polyacrylamide gel (Figure 2; size fractionation 1).
- 3’ linker II ligation
- Mix the digested DNA (from step J9) with 2 µl of 10 µM 3’ linker II and 1 µl of Quick T4 DNA ligase (NEB) in 1x Quick ligation buffer.
- Incubate the ligation reaction mixture at room temperature for 15 min, purify it using a QIAquick PCR Purification Kit, and elute it with 100 µl of TE buffer.
- Mix the digested DNA (from step J9) with 2 µl of 10 µM 3’ linker II and 1 µl of Quick T4 DNA ligase (NEB) in 1x Quick ligation buffer.
- Second PCR optimization (see Note 3)
- To determine the optimal number of PCR cycles, prepare a 0.2-ml PCR tube containing 5 µl of the ds cDNA ligated with 5’ linker I/3’ linker II (from step K2), 0.5 µl of 25 µM 5’ linker I forward primer, 0.5 µl of 25 µM 3’ linker II PCR primer, 5 µl of 1x Advantage 2 PCR buffer, 1 µl of 10 mM dNTP mix, 1 µl of 50x Advantage 2 polymerase mix, and MilliQ water in a 50 µl volume.
- Perform PCR with the following cycling parameters: 6 cycles of 98 °C for 10 sec and 68 °C for 10 sec. After the 6 cycles, transfer 5 μl of the reaction to a clean microcentrifuge tube.
- Perform PCR with the rest of the PCR reaction mixture for an additional 3 cycles of 98 °C for 10 sec and 68 °C for 10 sec.
- After these additional 3 cycles, transfer 5 μl of the reaction to a clean microcentrifuge tube.
- In the same way, repeat additional PCR cycles until 18 total cycles are reached.
- Thus, prepare a series of PCR reactions of 6, 9, 12, 15, and 18 cycles and analyze by 20% polyacrylamide gel electrophoresis to compare the band patterns (Figure 3; PCR optimization 2).
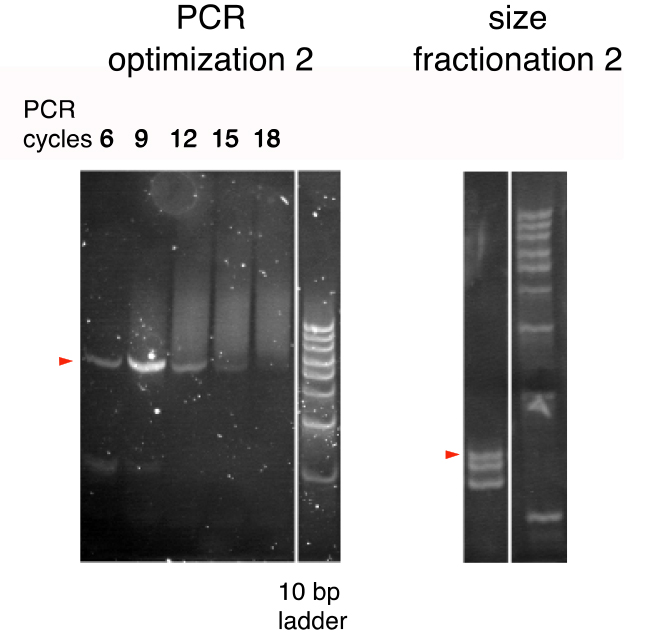
Figure 3. The second PCR cycle optimization and size fractionation (Arakawa, 2016). PCR products were run on 20% polyacrylamide gels. A 10-bp ladder was used as the size marker. Bands of the expected sizes are marked by triangles. (see Notes 4 and 5) - Determine the optimal number of PCR cycles as the minimal number of PCR cycles yielding the greatest quantity of the 72-bp product (typically around 9 cycles).
- Repeat five PCR reactions, each containing 50 µl, with the optimal number of PCR cycles.
- Purify the PCR product using a QIAquick PCR Purification Kit and elute it with 100 µl of TE buffer.
- To determine the optimal number of PCR cycles, prepare a 0.2-ml PCR tube containing 5 µl of the ds cDNA ligated with 5’ linker I/3’ linker II (from step K2), 0.5 µl of 25 µM 5’ linker I forward primer, 0.5 µl of 25 µM 3’ linker II PCR primer, 5 µl of 1x Advantage 2 PCR buffer, 1 µl of 10 mM dNTP mix, 1 µl of 50x Advantage 2 polymerase mix, and MilliQ water in a 50 µl volume.
- BsmBI/AatII digestion
- Digest the PCR product (from step L9) with 10 µl of BsmBI (10 U/µl, NEB) in 1x NEBuffer 3.1 in a 100 µl volume at 55 °C for 6 h, and then add 5 µl of AatII (20 U/µl, NEB) (see Note 2) to the solution; leave the solution at 37 °C overnight.
- Run the BsmBI/AatII digested DNA on a 20% polyacrylamide gel.
- Typically, 3 bands, corresponding to 25, 24, and 23 bp, are visible (Figure 2; size fractionation 2).
- Cut the 25-bp fragment out of the gel to purify by the crush and soak procedure as described in step K, and dissolve the fragment into 50 µl of TE buffer.
- Measure the concentration of the purified DNA by a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific).
- Digest the PCR product (from step L9) with 10 µl of BsmBI (10 U/µl, NEB) in 1x NEBuffer 3.1 in a 100 µl volume at 55 °C for 6 h, and then add 5 µl of AatII (20 U/µl, NEB) (see Note 2) to the solution; leave the solution at 37 °C overnight.
- Cloning
- Digest the lenti CRISPR ver. 2 (lentiCRISPR v2) (Sanjana et al., 2014) (Addgene) with BsmBI, treat with calf intestine phosphatase, extract with phenol/chloroform, and purify by ethanol precipitation.
- Mix 5 ng of the purified 25-bp guide sequence fragment (from step M5) with 3 µg of lentiCRISPR v2 and 1 µl of Quick T4 DNA ligase (NEB) in 1x Quick ligation buffer in a 40 µl volume.
- Incubate the ligation reaction mixture at room temperature for 15 min and then purify by ethanol precipitation.
- Electroporate the prepared gRNA library into STBL4 electro-competent cells (Invitrogen) using the following electroporator conditions: 1,200 V, 25 µF, and 200 Ω.
- Digest the lenti CRISPR ver. 2 (lentiCRISPR v2) (Sanjana et al., 2014) (Addgene) with BsmBI, treat with calf intestine phosphatase, extract with phenol/chloroform, and purify by ethanol precipitation.
- Deep sequencing
- To amplify the guide sequences, prepare a 0.2-ml PCR tube containing 1 µl of 100 ng/µl of lentiviral plasmid library (from step N4), 0.5 µl of 25 µM lentiCRISPR forward primer, 0.5 µl of 25 µM lentiCRISPR reverse primer, 5 µl of 1x Advantage 2 PCR buffer, 1 µl of 10 mM dNTP mix, 1 µl of 50x Advantage 2 polymerase mix, and MilliQ water in a 50 µl volume.
- Perform PCR with the following cycling parameters: 15 cycles of 98 °C for 10 sec and 68 °C for 10 sec.
- Purify the 100-bp PCR fragment containing the guide sequence from the 2% agarose gel using a QIAquick Gel Extraction Kit (QIAGEN). Prepare the deep sequencing library using a TruSeq Nano DNA Library Preparation Kit (Illumina).
- Deep sequence using Miseq (Illumina) (see Note 6).
- To amplify the guide sequences, prepare a 0.2-ml PCR tube containing 1 µl of 100 ng/µl of lentiviral plasmid library (from step N4), 0.5 µl of 25 µM lentiCRISPR forward primer, 0.5 µl of 25 µM lentiCRISPR reverse primer, 5 µl of 1x Advantage 2 PCR buffer, 1 µl of 10 mM dNTP mix, 1 µl of 50x Advantage 2 polymerase mix, and MilliQ water in a 50 µl volume.
Data analysis
- Demultiplex FASTQ files by Illumina Miseq.
- Trim the sequence reads to exclude vector backbone sequences using the CLC Genomics Workbench (QIAGEN) (see Note 6) and add the PAM-sequence NGG to the sequence reads.
- Align these sequence reads to a reference genome using the RNA-seq analysis toolbox in the CLC Genomics Workbench (QIAGEN).
Notes
- The quality of the poly(A) RNA is one of the most important factors that affects the library’s quality (Procedure B). During setup of the methodology for gRNA library construction, rRNA contamination was observed in poly(A) RNA purified using an oligodT column, and rRNA-originated guide sequences sometimes occupied 40-50% of the total original library. Since rRNA occupies more than 90% of intracellular RNA, generally speaking, it is hard to avoid having some rRNA contamination. The levels of rRNA contamination could be tested by the Bioanalyzer (Agilent). If a high amount of rRNA is contaminated, the washing step with buffer OBB (steps B7-B10) must be repeated. Alternatively, an rRNA depletion kit could be incorporated into the protocol to reduce rRNA contamination.
- PCR artifacts amplifying the linker sequences were also observed during setup of the methodology. For this reason, the linker sequence was designed with additional restriction sites, namely BglII for the 5’ SMART tag (step H3), XbaI for the 3’ linker I (step J1), and AatII for the 5’ linker I and 3’ linker II (step N1). By cutting with these additional restriction enzymes, it was possible to remove most of the PCR artifacts amplifying the linker sequences.
- Because the PCR conditions are optimized for Advantage 2 polymerase (Clontech), I recommend using Advantage 2, which is optimal for efficient amplification of a complex cDNA library (Procedure I and Procedure M). PCR conditions, such as cycling number, primer concentration, or template amount, have to be optimized if other polymerases are used for PCR.
- PCR cycle number has to be carefully titrated because the desired PCR products will be reduced or lost by over-cycling of PCR (Figures 2 and 3).
- The BsmBI restriction digest of the final PCR reaction generated the right size of DNA fragment (25 bp) in addition to one- or two-bp shorter, unexpected DNA fragments (Figure 3). These shorter DNA fragments were probably due to the inaccuracy of the cleavage position of the type III and type IIS restriction enzymes. The 25-bp fragment should be carefully isolated in order to avoid contamination of the 24- or 23-bp fragment, which may not have a PAM in the proper position.
- The deep sequencer and sequence analysis software can be chosen by users depending on their purpose and lab environment (Procedure P, Data analysis). For example, deep sequencing can also be done by the Illumina HiSeq.
Recipes
- TE buffer (10 mM Tris HCl [pH 8], 1 mM Na2EDTA)
For 100 ml:
1 ml 1 M Tris HCl (pH 8)
0.4 ml 250 mM Na2EDTA
98.6 ml MilliQ water
Autoclave and then store at room temperature
Acknowledgments
I am grateful to Giulia Bastianello for critical reading the manuscript. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. This protocol is adopted from Arakawa (2006).
References
- Arakawa, H. (2016). A method to convert mRNA into a gRNA library for CRISPR/Cas9 editing of any organism. Sci Adv 2(8): e1600699.
- Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A. and Horvath, P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819): 1709-1712.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Grissa, I., Vergnaud, G. and Pourcel, C. (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8: 172.
- Koike-Yusa, H., Li, Y., Tan, E. P., Velasco-Herrera Mdel, C. and Yusa, K. (2014). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32(3): 267-273.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Sanjana, N. E., Shalem, O. and Zhang, F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11(8): 783-784.
- Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., Heckl, D., Ebert, B. L., Root, D. E., Doench, J. G. and Zhang, F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166): 84-87.
- Wang, T., Wei, J. J., Sabatini, D. M. and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343(6166): 80-84.
- Zhou, X., Marks, P. A., Rifkind, R. A. and Richon, V. M. (2001). Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A 98(19): 10572-10577.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Arakawa, H. (2017). A Method to Convert mRNA into a Guide RNA (gRNA) Library without Requiring Previous Bioinformatics Knowledge of the Organism. Bio-protocol 7(10): e2319. DOI: 10.21769/BioProtoc.2319.
Category
Molecular Biology > DNA > Gene expression
Systems Biology > Genomics > Sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.