- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Formation of Minimised Hairpin Template-transcribing Dumbbell Vectors for Small RNA Expression


Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2313 Views: 10384
Reviewed by: Longping Victor TseGal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
A major barrier for using non-viral vectors for gene therapy is the short duration of transgene expression in postmitotic tissues. Previous studies showed transgene expression from conventional plasmid fell to sub-therapeutic level shortly after delivery even though the vector DNA was retained, suggesting transcription was silenced in vivo (Nicol et al., 2002; Chen et al., 2004). Emerging evidence indicates that plasmid bacterial backbone sequences are responsible for the transcriptional repression and this process is independent of CpG methylation (Chen et al., 2008). Dumbbell-shaped DNA vectors consisting solely of essential elements for transgene expression have been developed to circumvent these drawbacks. This novel non-viral vector has been shown to improve transgene expression in vitro and in vivo (Schakowski et al., 2001 and 2007). Here we describe a novel method for fast and efficient production of minimised small RNA-expressing dumbbell vectors. In brief, the PCR-amplified promoter sequence is ligated to a chemically synthesized hairpin RNA coding DNA template to form the covalently closed dumbbell vector. This new technique may facilitate applications of dumbbell-shaped vectors for preclinical investigation and human gene therapy.
Keywords: Dumbbell vectorBackground
With regard to delivery, a small vector size is advantageous improving extracellular transport including extravasation and diffusion through the extracellular matrix network as well as cellular uptake and nuclear diffusion. Various methods for dumbbell vector production have been developed over the time including methods for the generation of dumbbells expressing small RNAs such as small hairpin RNAs (shRNAs) and microRNAs (miRNAs) (Schakowski et al., 2001; Taki et al. 2004). These vectors usually harbour redundant sequences as the expressed RNAs are self-complementary. We eliminated redundant sequences generating minimised dumbbell vectors in which transcription goes around the hairpin structure of the dumbbell itself (Jiang et al., 2016). Such minimised dumbbell vectors can be as short as 130 bp representing the smallest expression vectors ever reported. An illustrated comparison between a conventional plasmid, a dumbbell harbouring a linear expression cassette, and a novel hairpin template-transcribing dumbbell vector is shown in Figure 1. This novel protocol facilitates the production of the new minimised small RNA expression dumbbell vectors.
.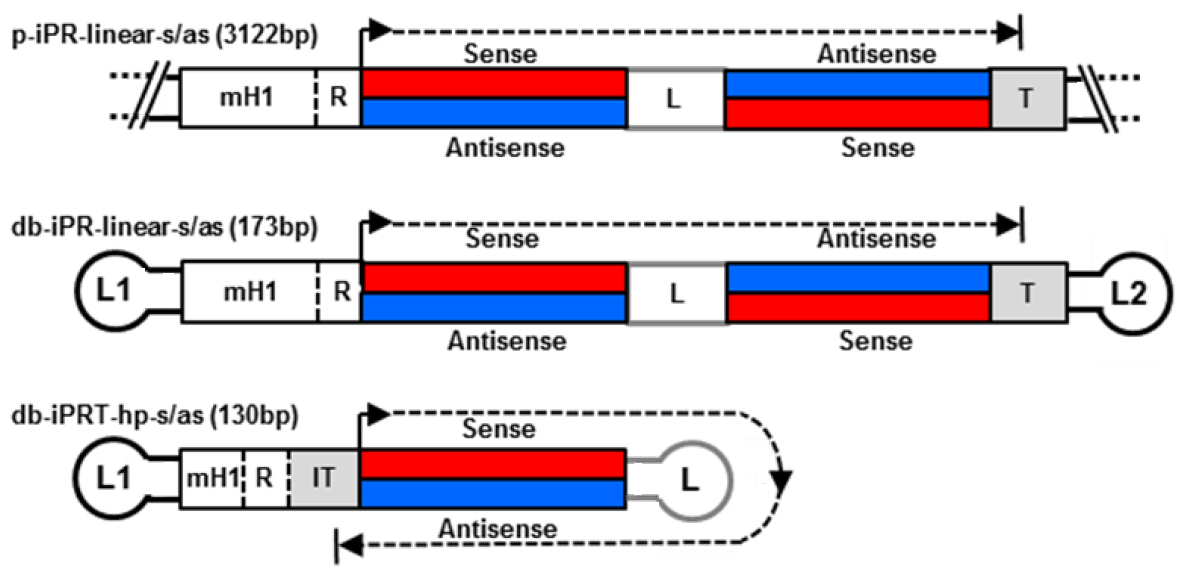
Figure 1. Structures of small hairpin RNA-expressing plasmid and dumbbell vectors. Upper two: conventional plasmid p-iPR-linear-s/as and dumbbell db-iPR-linear-s/as vectors with linear shRNA expression cassettes and integrated promoter-restriction endonuclease site element (iPR). Lower vector: minimized hairpin template (hp) dumbbell harboring an iPRT element. R indicates a restriction overhang ligation site. T indicates termination signal. IT indicates inverted termination signal. Loops L1 and L2 are (T)4 tetra loops.
Materials and Reagents
- 0.2-10 μl pipette tips, Corning® Isotip® filtered (Corning, catalog number: 4807 )
- 1-200 μl pipette tips (Corning, Axyge®, catalog number: TF-200-R-S )
- 100-1,000 μl pipette tips (Corning, Axygen®, catalog number: TF-1000-R-S )
- 1.5 ml microcentrifuge tubes (RNase, DNase and Pyrogen-Free) (Corning, Axygen®, catalog number: MCT-150-C )
- 0.2 ml thin-walled PCR tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3412 )
- Falcon® 50 ml conical centrifuge tubes (Corning, Falcon®, catalog number: 352070 )
- pSuper-basic vector (Oligoengine, catalog number: VEC-pBS-0002 )
- miRNA-coding oligonucleotide (PAGE purified) 5’-pGATCTAGCACGACTCGCAGCTCCCAAGAGCCTAACCCGTGGATTTAAACGGTAAACATCACAAGTTAGGGTCTCAGGGACTGAGAGGAGCGCAA-3’
- Oligonucleotides for minimal H1 (mH1) promoter (PAGE purified)
mH1-Fw: 5’-pAATTCATATTTGCATGTCGCTATGTGTTCTGGGAAATCACCATAAACGTGAAATGTCTTTGGATTTGGGAATCTTATAAGTTCTGTATGAGAGCACAGA-3’
mH1-Rv: 5’-pGATCTCTGTGCTCTCATACAGAACTTATAAGATTCCCAAATCCAAAGACATTTCACGTTTATGGTGATTTCCCAGAACACATAGCGACATGCAAATATG-3’ - UltraPureTM DNase/RNase-Free distilled water (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977015 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M2670-100G )
- dNTP set 100 mM solutions (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0181 )
- Oligonucleotide primers for mH1 promoter amplification (HPCL purified)
NbBpu-Fw
5’-pTTAGGAGTTTTCTCCTAAGCATATTTGCATGTCGCTATGTGTTCTG-3’
BamHI-Rv
5’-TGCAGGATCCCTGTGCTCTCATACAGAACTTATAAGATTCCC-3’ - Taq DNA polymerase, recombinant (5 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0402 )
- QIAquick PCR Purification Kit (QIAGEN, catalog number: 28106 )
- 10x FD buffer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: B64 )
- Nb.Bpu10I (5 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: ER1681 )
- FastDigest BamHI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0054 )
- shRNA-coding oligonucleotide (PAGE purified) 5’-pGATCTAAAAAGAGCTGTTTCTGAGGAGCCTCTCTTGAAGGCTCCTCAGAAACAGCTCTTTTTA-3’
- T4 DNA ligase (5 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EL0014 )
- Neutralizing oligonucleotide (HPLC purified)
5’-TTAGGAGTTTTCTCCTAA-3’ - Adenosine 5’-triphosphate disodium salt hydrate (Sigma-Aldrich, catalog number: A2383-1G )
- FastDigest BglII (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0083 )
- T7 DNA polymerase (10 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0081 )
- Phenol solution (Sigma-Aldrich, catalog number: P4557-100ML )
- Chloroform (Sigma-Aldrich, catalog number: 288306-1L )
- 3-methyl-1-butanol (Sigma-Aldrich, catalog number: 309435-100ML )
- Ethanol, absolute (Fisher Scientific, catalog number: BP28184 )
- 3 M potassium acetate (pH 4.8)
- Sodium acetate (Sigma-Aldrich, catalog number: S2889-250G )
- FastDigest EcoRI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0274 )
- Agarose, LE, analytical grade (Promega, catalog number: V3125 )
- Ethidium bromide solution (Bio-Rad Laboratories, catalog number: 1610433 )
- GeneRuler DNA ladder mix (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM0331 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888-500G )
- Tris-HCl (Powder) (Roche Diagnostics, catalog number: 10812846001 )
- EDTA (Sigma-Aldrich, catalog number: EDS-100G )
- 10x hybridization buffer (see Recipes)
- TE buffer (see Recipes)
Equipment
- Pipettes (Gilson, PIPETMAN® Classic, models: P2, P20N, P200N, and P1000N )
- Standard thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, model: GeneAmp PCR System 9700 )
Note: This product has been discontinued. - Gel doc (Bio-Rad Gel Doc Imager)
- Gel running apparatus (Thermo Fisher Scientific, Amersham BiosciencesTM)
- Gel staining tray
- Benchtop centrifuge (Eppendorf, model: 5430 R )
- Heat block (Eppendorf, model: Thermomixer® Comfort )
- Spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDrop 2000 )
- Glass beaker (Schott, Duran)
- Microwave (Panasonic)
Software
- ImageJ (http://imagej.nih.gov/ij/)
Procedure
In this protocol we describe a method for the production of a) shRNA (example db-iPRT-hp-s/as: targeting luciferase) or b) miRNA (example db-hp-miR-125b-1: expressing has-miR-125b-1) expressing minimised dumbbell vectors (Jiang et al., 2016). For these dumbbells, shRNA or miRNA expression is driven by the human minimal H1 (mH1) promoter. The mH1 promoter is PCR-amplified using a forward primer which introduces a cleavage site for a nicking endonuclease and a reverse primer introducing a cleavage site for a conventional restriction endonuclease. After incubating the PCR product with both endonucleases, one loop structure of the dumbbell is formed by the refolding overhang generated by the nicking reaction, whereas the other loop is formed by ligation of a shRNA or miRNA-coding hairpin oligodeoxyribonucleotide. Ligation is performed in the presence of appropriate restriction enzymes to suppress the formation of misligated products (Cost, 2007). The enzymatic ligation assisted by nucleases (ELAN) method suppresses the formation of misligated products such as dimers composed of the loop oligos or the expression cassette which are being cleaved in the presence of restriction endonuclease (in this example BamHI and BglII), thereby facilitating the formation of the intended dumbbell structure which doesn’t comprise the respective endonuclease cleavage sites. Finally, non-ligated DNA is destroyed by exonuclease treatment and exonuclease-resistant dumbbells are purified (Figure 2).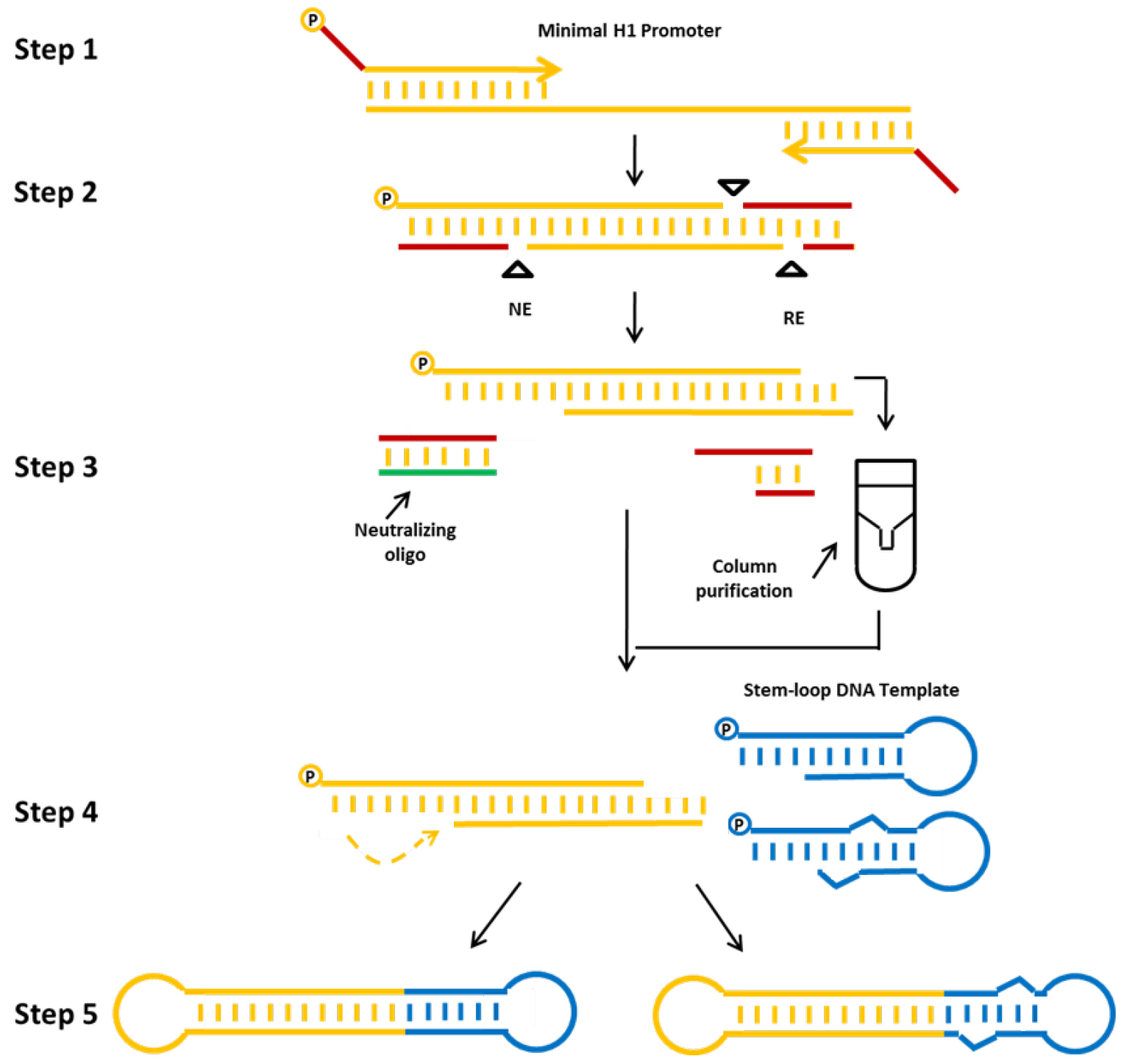
Figure 2. Production strategy for small RNA-expressing minimized dumbbell vectors. The protocol consists of the following steps: First, the mH1 promoter sequence is amplified by PCR. Proper nicking enzyme (NE) and conventional restriction (RE) sites are introduced via the PCR primers. Second, the amplified promoter DNA is digested using the corresponding nicking and restriction enzymes. Third, the digested DNA is purified, and fourth annealed and ligated with the hairpin DNA template oligo using T4 DNA ligase to form an shRNA (left side) or miRNA (right side) expressing dumbbell vector. The addition of a neutralizing oligo and the column purification step significantly improve dumbbell yields. Finally, non-ligated DNA is removed by exonuclease treatment and dumbbell vector DNA is purified using standard DNA purification techniques (Jiang et al., 2016).
- Annealing of oligonucleotides
- Correct folding of sh/miRNA-coding DNA hairpins is achieved by heating 500 pmol oligo DNA to 95 °C for 3 min (Thermomixer Comfort, Eppendorf) in 10 μl of 1x hybridization buffer and letting the solution cool down to room temperature within 1 h.
- The mH1 promoter in this protocol has been modified by introduction of an inverted transcriptional terminator. Therefore, the oligonucleotides mH1-Fw and mH1-Rv are dissolved in nuclease-free distilled water to a concentration of 100 μM. 500 pmol of each strand are annealed in 10 μl 1x hybridization buffer by heating to 95 °C for 3 min (Thermomixer Comfort, Eppendorf) followed by cooling down the solution to room temperature during 1 h (Note 1).
- Correct folding of sh/miRNA-coding DNA hairpins is achieved by heating 500 pmol oligo DNA to 95 °C for 3 min (Thermomixer Comfort, Eppendorf) in 10 μl of 1x hybridization buffer and letting the solution cool down to room temperature within 1 h.
- PCR amplification of the mH1 promoter sequence
- Add the following components into a thin-walled PCR tube:

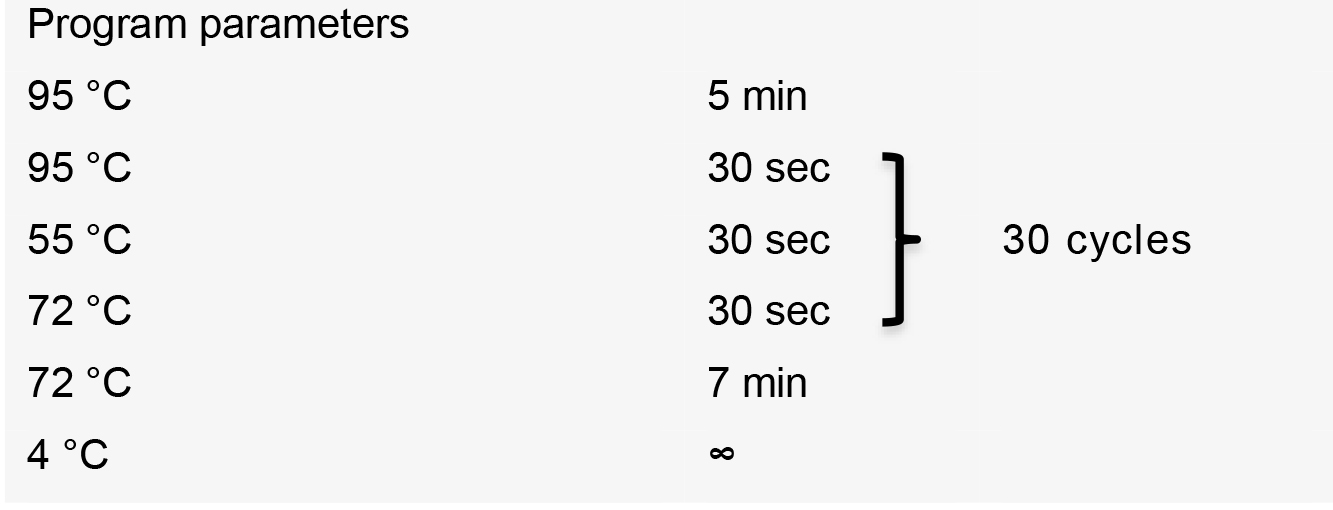
- Perform PCR using the following thermal cycler conditions:
PCR product is purified using QIAquick PCR Purification Kit (QIAGEN) following the manufacturer’s recommendations.
- Add the following components into a thin-walled PCR tube:
- Endonucleolytic cleavage of the PCR product
- Combine the reaction components at room temperature in the order indicated:

- Incubate the reaction mixture at 37 °C for 4 h, then 85 °C for 5 min to inactivate the enzymes (Note 2).
- Digested DNA is then purified with PCR QIAquick PCR Purification Kit (QIAGEN) following the manufacturer’s recommendations (Note 3).
- Combine the reaction components at room temperature in the order indicated:
- Annealing and ligation of the mH1 promoter and the sh/miRNA-coding hairpin oligonucleotide
- Annealing of promoter DNA and sh/miRNA-coding hairpin oligonucleotide is achieved by heating equimolar amounts (80 pmol) of each DNAs in 1x hybridization buffer to 95 °C for 3 min (Thermomixer Comfort, Eppendorf) and letting the solution cool down to room temperature during 1 h.
- After annealing, the ligation reaction is set up in the indicated order (Note 4):

- Incubate the reaction mixture at 22 °C for 4 h or overnight, then incubate at 80 °C for 10 min to inactivate the enzymes (Note 5).
- Withdraw a sample (1-2 µl) from the reaction mixture for gel electrophoresis analysis.
- Annealing of promoter DNA and sh/miRNA-coding hairpin oligonucleotide is achieved by heating equimolar amounts (80 pmol) of each DNAs in 1x hybridization buffer to 95 °C for 3 min (Thermomixer Comfort, Eppendorf) and letting the solution cool down to room temperature during 1 h.
- Exonuclease treatment and purification of the dumbbell-shaped vector
- Add 1 µl of T7 DNA polymerase (10 U/µl) to the 200 μl ligation mix above, incubate at 37 °C for 1 h and then at 80 °C 10 min to inactivate the enzyme (Note 6).
- Withdraw a sample (1-2 µl) from the reaction mixture for gel electrophoresis analysis.
- Perform analytical 1.5% agarose gel electrophoresis of the withdrawn samples to monitor the conversion yields and purity of dumbbell vector DNA (Figure 3).
- Purify the dumbbell DNA using standard phenol/chloroform extraction followed by ethanol precipitation. In detail, add an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) to the aqueous dumbbell solution, vortex for 30 sec, and separate the aqueous and organic phase by centrifugation (5 min, 13,000 x g). Transfer the upper aqueous phase to a new Eppendorf tube and re-extract residues of phenol. Therefore, add an equal volume of chloroform/isoamyl alcohol (24:1), shake rigorously by hand for 30 sec, and separate the phase by centrifugation (30 sec, 13,000 x g). Transfer the upper aqueous phase to a new Eppendorf tube and repeat the re-extraction process twice (Note 7). For ethanol precipitation, the aqueous phase was topped up to 400 µl with distilled water, then 0.1 volumes (40 µl) of 3 M potassium acetate (pH 4.8) followed by 2.5 volumes (1,100 µl) ethanol were added, the solution was mixed and incubated at -20 °C for 20 min. DNA was pelleted by centrifugation at 13,000 x g, 4 °C for 15 min. The pellets were then air-dried. Dissolve the purified DNA in TE buffer or distilled water.
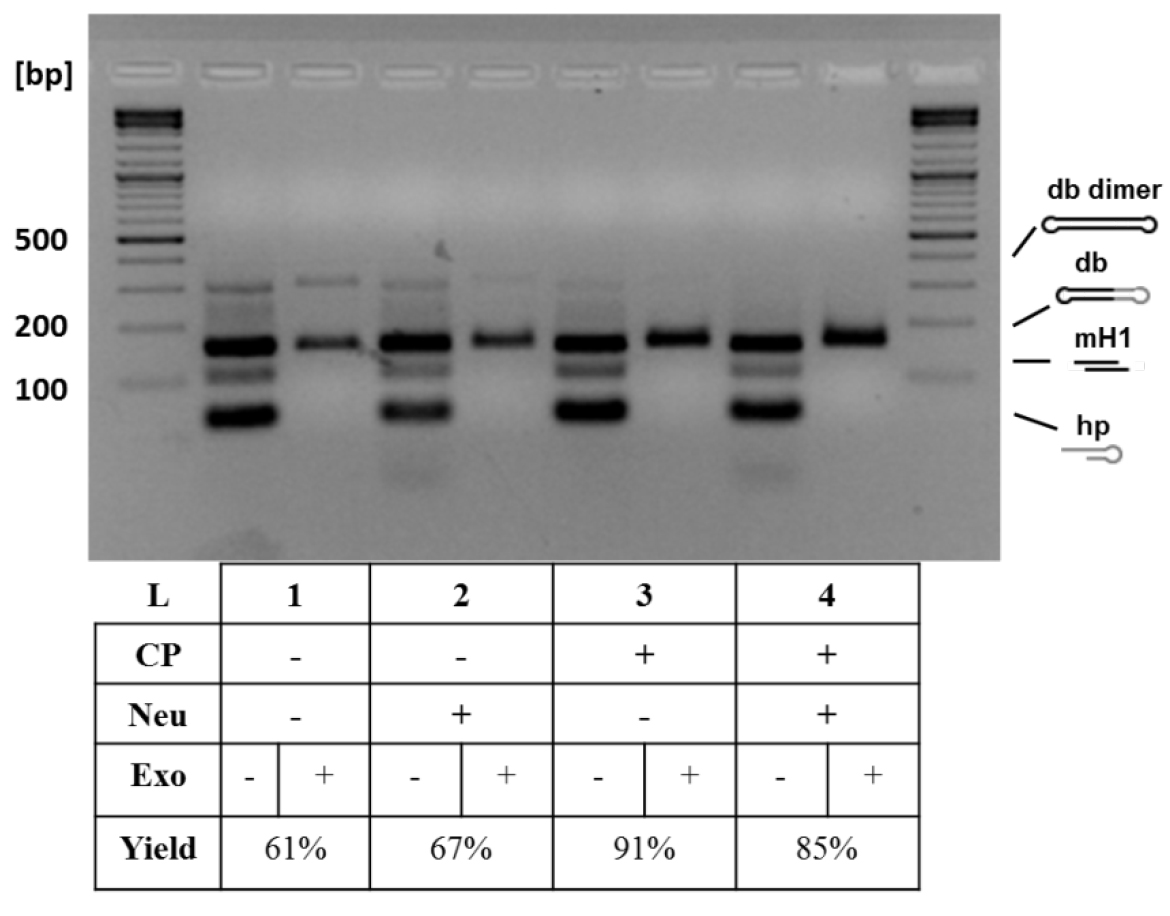
Figure 3. Analytical gel electrophoresis of dumbbell vector DNA after different treatments. Additional treatments increased dumbbell conversion yield: basic protocol (1), basic protocol with neutralizing oligo (50 pmol) (2), basic protocol with column purification (using the QIAquick PCR Purification Kit and following standard protocol by QIAGEN) (3), and basic protocol with both treatments (4). The neutralizing oligo binds to the oligo released after nicking enzyme cleavage and prevents that this can bind again to the generated overhang. The column purification removes the small cleavage products and prevents them from relegation in the subsequent ligation step. The highest conversion yield (91%) was achieved with the addition of column purification step. The conversion yield is defined as the yield obtained when comparing the size of the expected dumbbell before and after exonuclease treatment. CP, column purification; Neu, neutralizing oligo (Jiang et al., 2016).
- Add 1 µl of T7 DNA polymerase (10 U/µl) to the 200 μl ligation mix above, incubate at 37 °C for 1 h and then at 80 °C 10 min to inactivate the enzyme (Note 6).
Data analysis
Analysis of dumbbell DNA conversion yield:
- Measure the intensity of the DNA bands corresponding to the dumbbell products in the electrophoresis gel with ImageJ using the wand tool.
- Conversion yield is calculated by dividing the intensity of the band after exonuclease treatment by that of the corresponding band in the ligation sample.
Notes
- Alternatively, the mH1 sequence can be generated through PCR amplification from plasmid DNA pSuper-mH1 which was constructed by cloning the hybridized oligonucleotides into the pSuper-basic plasmid. To insert the annealed sequence into the pSuper-basic plasmid, the vector (1 µg) was first digested with EcoRI and BglII at 37 °C for 4 h. After digestion, the DNA was purified using the QIAquick PCR Purification Kit. Ligation was performed using equimolar amounts (0.25 pmol) of the digested vector backbone and annealed insert at 22 °C for 4 h. The correct clone was confirmed by sequencing. If pSuper-mH1 was used for the PCR reaction, 10 ng was used as the template.
- We used Nb.Bpu10I as the nicking enzyme based on a previous report by Taki et al. (2004). The enzyme is supplied with 10x buffer R but here the universal 10x buffer FD was used instead after consulting Thermo Fisher’s technical support.
- Although the digestion product from this step can directly be used for ligation, we found an additional purification step could greatly increase the dumbbell conversion yield as shown in Figure 2.
- In some cases, a neutralising oligo was added into the ligation reaction to suppress reannealing of the short nicking fragment (see Figure 2). Ligation was performed in FD buffer complemented with 1 mM final ATP instead of the ligation buffer since all enzymes including the ligase were 100% active in this buffer. To ligate the sh/miRNA-coding hairpin template and the promoter DNA, we followed the enzymatic ligation assisted by nucleases (ELAN) technique as described before (Cost, 2007). The sh/miRNA-coding hairpin oligonucleotides are ordered PAGE purified and can directly be used for ligation.
- According to our experience, the ligation reaction was completed within 4 h and longer incubation times did not improve the dumbbell yield.
- T7 DNA polymerase exhibits 100% activity in the FD buffer and was therefore directly added into the ligation mixture.
- It is essential to shake by hand instead of vortexing in order to mix the phase efficiently.
Recipes
- 10x hybridization buffer
1 M NaCl
100 mM MgCl2
200 mM Tris-HCl, pH 7.4 - TE buffer
10 mM Tris-HCl, pH 8.0
1 mM EDTA
Acknowledgments
The protocol described herein was developed and utilized previously in Jiang et al. (2016). This work was supported by the National University of Singapore [Bridging Grant NUHSRO/2015/091/Bridging/02], the National Medical Research Council of Singapore [New Investigator Grant number NMRC/NIG/1058/2011], and the Ministry of Education of Singapore [Academic Research Fund (AcRF) Tier 1 Faculty Research Committee (FRC) grants number T1-2011Sep-04 and T1-2014Apr-02 and Seed Fund for Basic Science Research number T1-BSRG 2015-05], all to VP. The authors declare competing financial interests. A patent application covering major parts of the work is pending.
References
- Chen, Z. Y., He, C. Y., Meuse, L. and Kay, M. A. (2004). Silencing of episomal transgene expression by plasmid bacterial DNA elements in vivo. Gene Ther 11(10): 856-864.
- Chen, Z. Y., Riu, E., He, C. Y., Xu, H. and Kay, M. A. (2008). Silencing of episomal transgene expression in liver by plasmid bacterial backbone DNA is independent of CpG methylation. Mol Ther 16(3): 548-556.
- Cost, G. J. (2007). Enzymatic ligation assisted by nucleases: simultaneous ligation and digestion promote the ordered assembly of DNA. Nat Protoc 2(9): 2198-2202.
- Jiang, X., Yu, H., Teo, C. R., Tan, G. S., Goh, S. C., Patel, P., Chua, Y. K., Hameed, N. B., Bertoletti, A. and Patzel, V. (2016). Advanced design of dumbbell-shaped genetic minimal vectors improves non-coding and coding RNA expression. Mol Ther 24(9): 1581-1591.
- Nicol, F., Wong, M., MacLaughlin, F. C., Perrard, J., Wilson, E., Nordstrom, J. L. and Smith, L. C. (2002). Poly-L-glutamate, an anionic polymer, enhances transgene expression for plasmids delivered by intramuscular injection with in vivo electroporation. Gene Ther 9(20): 1351-1358.
- Schakowski, F., Gorschluter, M., Buttgereit, P., Marten, A., Lilienfeld-Toal, M. V., Junghans, C., Schroff, M., Konig-Merediz, S. A., Ziske, C., Strehl, J., Sauerbruch, T., Wittig, B. and Schmidt-Wolf, I. G. (2007). Minimal size MIDGE vectors improve transgene expression in vivo. In Vivo 21(1): 17-23.
- Schakowski, F., Gorschluter, M., Junghans, C., Schroff, M., Buttgereit, P., Ziske, C., Schottker, B., Konig-Merediz, S. A., Sauerbruch, T., Wittig, B. and Schmidt-Wolf, I. G. (2001). A novel minimal-size vector (MIDGE) improves transgene expression in colon carcinoma cells and avoids transfection of undesired DNA. Mol Ther 3(5 Pt 1): 793-800.
- Taki, M., Kato, Y., Miyagishi, M., Takagi, Y. and Taira, K. (2004). Small-interfering-RNA expression in cells based on an efficiently constructed dumbbell-shaped DNA. Angew Chem Int Ed Engl 43(24): 3160-3163
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jiang, X. and Patzel, V. (2017). Formation of Minimised Hairpin Template-transcribing Dumbbell Vectors for Small RNA Expression. Bio-protocol 7(11): e2313. DOI: 10.21769/BioProtoc.2313.
Category
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.