- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Endogenous C-terminal Tagging by CRISPR/Cas9 in Trypanosoma cruzi



(*contributed equally to this work) Published: Vol 7, Iss 10, May 20, 2017 DOI: 10.21769/BioProtoc.2299 Views: 15799
Reviewed by: Renate WeizbauerJingyu PengAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
To achieve the C-terminal tagging of endogenous proteins in T. cruzi we use the Cas9/pTREX-n vector (Lander et al., 2015) to insert a specific tag sequence (3xHA or 3xc-Myc) at the 3’ end of a specific gene of interest (GOI). Chimeric sgRNA targeting the 3’ end of the GOI is PCR-amplified and cloned into Cas9/pTREX-n vector. Then a DNA donor molecule to induce DNA repair by homologous recombination is amplified. This donor sequence contains the tag sequence and a marker for antibiotic resistance, plus 100 bp homology arms corresponding to regions located right upstream of the stop codon and downstream of the Cas9 target site at the GOI locus. Vectors pMOTag23M (Oberholzer et al., 2006) or pMOHX1Tag4H (Lander et al., 2016b) are used as PCR templates for DNA donor amplification. Epimastigotes co-transfected with the sgRNA/Cas9/pTREX-n construct and the DNA donor cassette are then cultured for 5 weeks with antibiotics for selection of double resistant parasites. Endogenous gene tagging is finally verified by PCR and Western blot analysis.
Keywords: CRISPR/Cas9Background
Genetic manipulation of protist parasites has significantly increased since the emergence of the CRISPR/Cas9 technology (Lander et al., 2016a). Trypanosoma cruzi is the causative agent of Chagas disease, which affects millions of people worldwide, particularly in Central and South America where the disease is endemic. Vaccines to prevent this disease have not been developed, and available drug treatments are not completely effective (Urbina and Docampo, 2003). This parasite has been particularly refractory to genetic manipulation (Docampo, 2011). However, the recent use of the CRISPR/Cas9 technology for gene knockout and knockdown (Peng et al., 2014; Lander et al., 2015) and to perform endogenous gene tagging (Lander et al., 2016b) has transformed the approaches for functional study of proteins in this organism. Here we describe a protocol to generate CRISPR/Cas9-mediated endogenous gene tagging in T. cruzi, leading to the expression of specific C-terminal tagged proteins in this parasite. Tagged proteins can be detected by Western blot analysis and their subcellular localization can be determined by immunofluorescence microscopy. Other potential applications of the technique include immunoprecipitation assays and tandem affinity purification (TAP) to establish protein-protein interactions.
Materials and Reagents
- Pipette tips for P10, P20, P200 and P1000 pipettes
- Microcentrifuge tubes (1.5 ml)
- 0.6 ml tube
- PCR tubes (0.2 ml)
- Petri dishes
- T25 culture flasks
- Centrifuge tubes (15 ml)
- Electroporation cuvettes 0.4 cm gap (Bio-Rad Laboratories, catalog number: 1652081 )
- Cas9/pTREX-n vector (Addgene, catalog number: 68708 ) (Lander et al., 2015)
- pUC_sgRNA vector (Addgene, catalog number: 68710 ) (Lander et al., 2015)
- pMOTag23M vector (Oberholzer et al., 2006)
- pMOHX1Tag4H vector (Lander et al., 2016b)
- Chemically competent E. coli DH5α cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18265017 ) (storage temperature: -80 °C)
- T. cruzi Y strain
- Platinum® Taq DNA polymerase high fidelity (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11304011 )
- Miniprep kit (Wizard® Plus SV Minipreps DNA Purification System) (Promega, catalog number: A1460 )
- DNA extraction kit from agarose gels (Wizard® SV Gel and PCR Clean-Up System) (Promega, catalog number: A9281 )
- BamHI (New England Biolabs, catalog number: R0136S ) (storage temperature: -20 °C)
- Antarctic Phosphatase (New England Biolabs, catalog number: M0289S ) (storage temperature: -20 °C)
- Agarose (Promega, catalog number: V3125 )
- 1 kb plus ladder
- T4 DNA ligase (Promega, catalog number: M1801 ) (storage temperature: -20 °C)
- 2x rapid ligation buffer (Promega, catalog number: C6711 ) (storage temperature: -20 °C)
- LB broth (Sigma-Aldrich, catalog number: L3022 )
- LB broth with agar (Sigma-Aldrich, catalog number: L2897 )
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A0166 )
- GoTaq G2 Flexi DNA polymerase (Promega, catalog number: M7805 ) (storage temperature: -20 °C)
- Ethanol
- DNA oligonucleotides, purity desalted (Exxtend Biotecnologia Ltda., Campinas, Brazil)
- DNA ultramer oligonucleotides, purity HPLC (Exxtend Biotecnologia Ltda., Campinas, Brazil)
- Acetic acid
- Phenol/chloroform/isoamyl alcohol (25:24:1)
- Heat inactivated fetal bovine serum (FBS) (Vitrocell Embriolife, catalog number: S0011) (storage temperature: -20 °C)
- Penicillin (10,000 U/ml)/streptomycin (10 mg/ml) stock solution (Vitrocell Embriolife, Campinas, Brazil) (storage temperature: -20 °C)
- Phosphate buffer saline (PBS) pH 7.4
- G418 disulfate (KSE Scientific, catalog number: 6483-TB-2G-001-5 )
- Puromycin dihydrochloride (Thermo Fisher Scientific, GibcoTM, catalog number: A1113803 )
- Hygromycin B (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10687010 )
- Anti-HA high affinity rat monoclonal antibody (clone 3F10) (Roche Diagnostics, catalog number: 11867423001 )
- Monoclonal anti-α-Tubulin antibody produced in mouse (clone B-5-1-2) (Sigma-Aldrich, catalog number: T5168 )
- HA epitope tag monoclonal antibody (clone 2-2.2.14) (Thermo Fisher Scientific, Invitrogen, catalog number: 26183 )
- Rabbit anti-HA polyclonal antibody (Y-11) (Santa Cruz Biotechnology, catalog number: sc-805 )
- Anti-c-Myc monoclonal antibody (clone 9E10) (Santa Cruz Biotechnology, catalog number: sc-40 )
- Rabbit anti-c-Myc polyclonal antibody (N-262) (Santa Cruz Biotechnology, catalog number: sc-764 )
- Dimethyl sulfoxide (DMSO)
- Sodium hydroxide (NaOH)
- Sodium chloride (NaCl)
- Potassium chloride (KCl)
- Sodium phosphate dibasic (Na2HPO4)
- D-(+)-glucose (Sigma-Aldrich, catalog number: G7021 )
- Liver infusion broth (BD, Difco, catalog number: 226920 )
- TrypticaseTM peptone (BD, BBL, catalog number: 211921 )
- Hemin (Sigma-Aldrich, catalog number: H9039 )
- Calcium chloride (CaCl2)
- Dibasic potassium phosphate (K2HPO4)
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- Ethylenediaminetetraacetic acid (EDTA)
- Tris base
- SOC medium (Sigma-Aldrich, catalog number: S1797 )
- Sodium acetate
- sgRNA amplification PCR reaction mix for 1 reaction (see Recipes)
- Colony PCR reaction mix for 1 reaction (see Recipes)
- Donor DNA PCR reaction mix for 1 reaction (see Recipes)
- Hemin stock solution (see Recipes)
- LIT medium (Liver Infusion Tryptose) (see Recipes)
- Electroporation buffer (Cytomix), pH 7.6 (see Recipes)
- 1x TAE (Tris-acetate-EDTA) buffer, pH 8.3 (see Recipes)
Equipment
- Pipettes (Gilson, Pipetman®, P10, P20, P200 and P1000)
- Incubator with refrigeration (28 °C) (Shel Lab, model: SSI5R )
- Incubator with agitation (37 °C) (Gallemkamp, model: IOI400.XX2.C )
- Electrophoresis chamber (Bio-Rad Laboratories, model: Mini-Sub® Cell GT Cell )
- Image digitalizer (UVITEC, model: Alliance 2.7 )
- NanoDrop spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000c )
- Microcentrifuge (Eppendorf, model: 5418 )
- Refrigerate centrifuge (Eppendorf, model: 5810 R )
- Electroporator (Bio-Rad Laboratories, model: Gene Pulser XcellTM Electroporation System )
- Neubauer chamber (Sigma-Aldrich, model: Bright-LineTM Hemacytometer, catalog number: Z359629 )
- Biological safety cabinet (Labconco, model: Class II A2, Purifier Cell Logic+ )
- Autoclave
Software
- DNAMAN software version 7.212 (Lynnon Corporation)
- Alliance 1D software (UVITEC)
Procedure
- Protospacer selection
- Search for the genomic DNA sequence of the T. cruzi gene of interest on TriTrypDB database (http://tritrypdb.org/tritrypdb/) and download that sequence plus 200 nucleotides downstream of the stop codon using the sequence retrieval tool.
- Select the protospacer region targeting the 3’end of the gene, to induce a double strand break by Cas9 nuclease in a site close to the stop codon (from 20 nt upstream of the stop codon to 50 nt downstream of the stop codon). The protospacer region is the 20-nt base-pairing sequence on the sgRNA. To target the template DNA strand, search in the transcribed gene sequence for 5’-N(20)-NGG-3’. The N(20) corresponds to the protospacer. NGG is the protospacer adjacent motif (PAM) sequence that is recognized by the Cas9 endonuclease. Alternatively, to target the non-template DNA strand, search in the transcribed gene sequence for 5’-CCN-N(20)-3’. The reverse complementary sequence of the N(20) will be used as the protospacer. Select the closest protospacer to the stop codon (preferably downstream of the open reading frame of the target gene) and verify that it does not generate Cas9 off-targeting on the T. cruzi genome. This can be done using online resources like EuPaGDT (Eukaryotic Pathogen CRISPR guide RNA Design Tool, http://grna.ctegd.uga.edu/) or specific scripts designed for this purpose (Sidik et al., 2014). Figure 1 shows an example of protospacer selection for gene tagging of TcVP1 (T. cruzi vacuolar proton pyrophosphatase coding sequence).
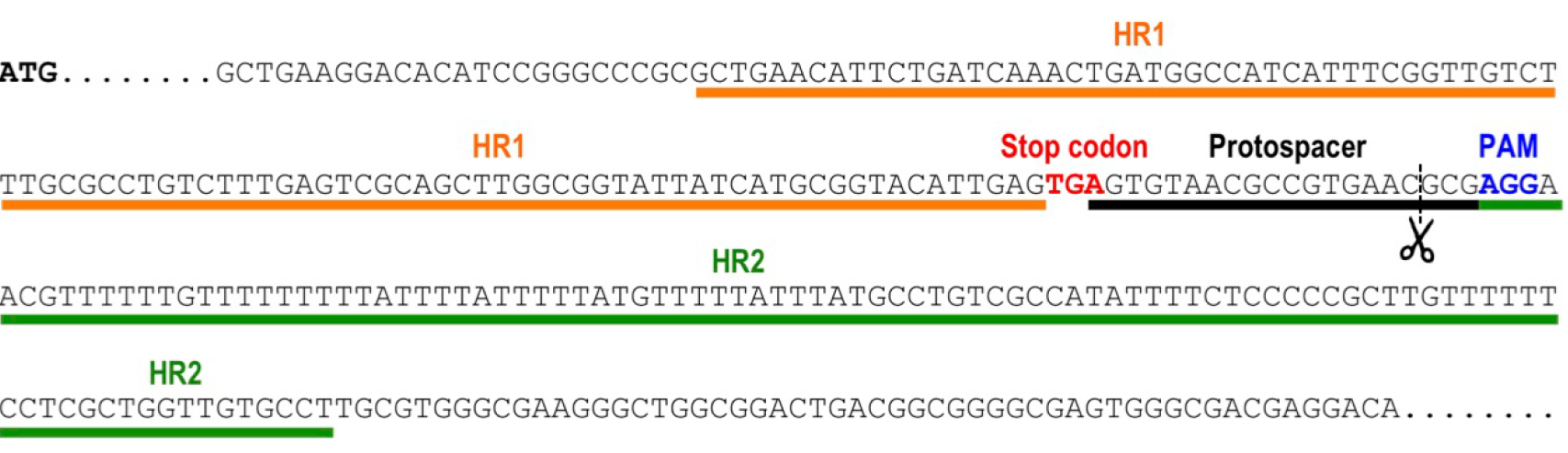
Figure 1. Selection of protospacer and homology regions for endogenous tagging of TcVP1 (TriTrypDB gene ID: TcCLB.510773.20). The schematic representation shows the genomic DNA sequence of TcVP1, including the start codon (ATG, bold sequence), the stop codon (TGA, red sequence), the chosen protospacer underlined in black, the protospacer adjacent motif (PAM, blue sequence) and the homology regions 1 (HR1) and 2 (HR2) underlined in orange and green, respectively, that were included in the DNA donor molecule to induce DNA repair by homologous recombination. Scissors indicate the Cas9 cut site.
- Search for the genomic DNA sequence of the T. cruzi gene of interest on TriTrypDB database (http://tritrypdb.org/tritrypdb/) and download that sequence plus 200 nucleotides downstream of the stop codon using the sequence retrieval tool.
- sgRNA amplification
- Design a specific forward primer for each sgRNA to be amplified, inserting the protospacer sequence N(20) into the following primer backbone: 5’-GATCGGATCC-N(20)-GTTTTAGAGCTAGAAATAGC-3’. The common reverse primer sequence to amplify the sgRNA is: 5’-CAGTGGATCCAAAAAAGCACCGACTCGGTG-3’. Each one of these primers contains a BamHI restriction site indicated in bold.
- Amplify sgRNA by PCR using the specific forward primer designed, the common reverse primer, and plasmid pUC_sgRNA as template (Recipe 1). PCR conditions using Platinum® Taq DNA polymerase high fidelity:
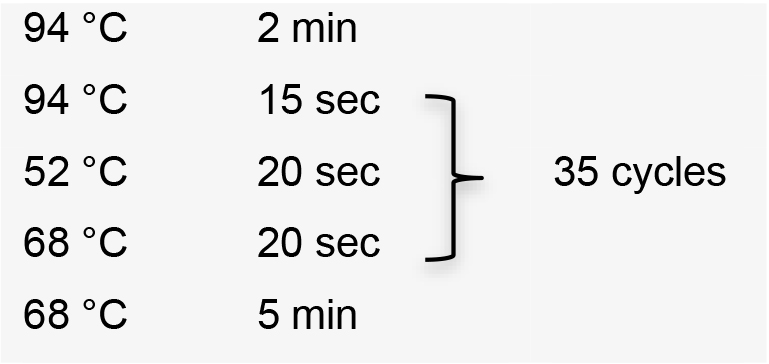
Hold temperature at 10 °C. Product PCR size: 122 bp. - Clean up PCR product using Wizard® SV Gel and PCR Clean-Up System.
- Design a specific forward primer for each sgRNA to be amplified, inserting the protospacer sequence N(20) into the following primer backbone: 5’-GATCGGATCC-N(20)-GTTTTAGAGCTAGAAATAGC-3’. The common reverse primer sequence to amplify the sgRNA is: 5’-CAGTGGATCCAAAAAAGCACCGACTCGGTG-3’. Each one of these primers contains a BamHI restriction site indicated in bold.
- sgRNA cloning into Cas9/pTREX-n vector
- Digest 1 μg of insert DNA (sgRNA purified PCR product) and 1 μg of DNA vector (Cas9/pTREX-n) separately with BamHI restriction enzyme (20 U) by incubating overnight at 37 °C.
- Dephosphorylate the digested vector by adding 5 U of Antarctic Phosphatase and 10x Antarctic Phosphatase buffer (final concentration: 1x) and incubate at 37 °C for 15 min. Immediately inactivate the phosphatase incubating the reaction for 5 min at 70 °C.
- Resolve digestion products by electrophoresis using 1.5% preparative agarose gel in 1x TAE buffer, and extract DNA from bands corresponding to insert (sgRNA, 122 bp) and vector (Cas9/pTREX-n, 11,173 bp) using Wizard® SV Gel and PCR Clean-Up System (Figure 2).
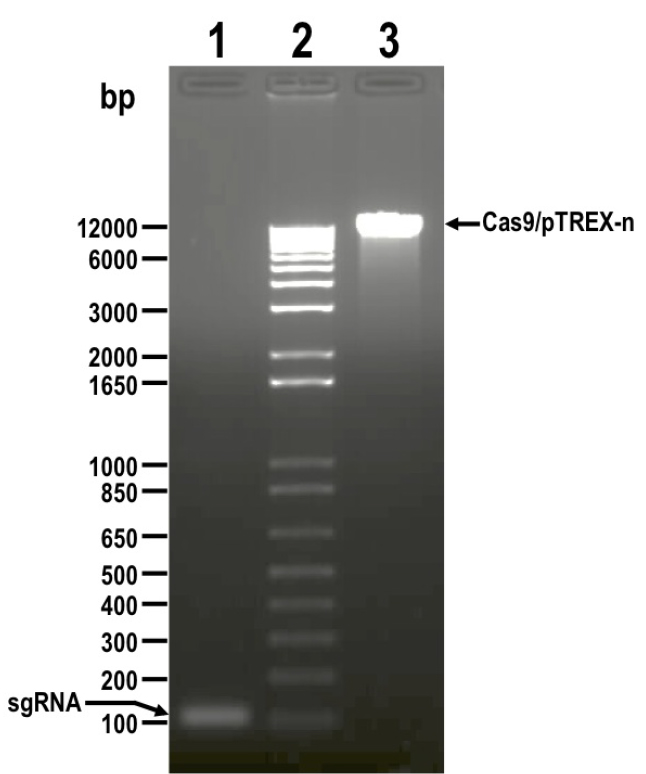
Figure 2. DNA fragments digested with BamHI restriction enzyme and resolved by electrophoresis in 1.5% agarose gel before DNA extraction. Lanes: 1) sgRNA, 2) 1 kb plus ladder, 3) Cas9/pTREX-n linearized vector. Molecular weights are shown on the left side. Fragments to be extracted are indicated with arrows. - Quantify isolated DNA fragments using a NanoDrop spectrophotometer.
- Perform ligation reaction by mixing the following reagents in a 0.6 ml tube: 100 ng vector and 22 ng insert (20:1 insert to vector molar ratio), T4 DNA ligase (3 U), 2x rapid ligation buffer (1/2 of the final reaction volume) and completing the final volume to 10 μl with ultrapure water. Incubate the reaction overnight at 4 °C.
- Use 5 μl of the ligation product to transform chemically competent DH5α E. coli cells following standard heat-shock transformation protocol.
- Spread cells on LB-agar plates supplemented with 100 μg/ml ampicillin and incubate overnight at 37 °C.
- Perform colony PCR in 0.2 ml tubes by manually picking individual colonies and resuspending them in 5 μl ultrapure water as template. Use the specific forward primer designed to amplify sgRNA, and the HX1 reverse primer 5’-TAATTTCGCTTTCGTGCGTG-3’ (Recipe 2). Colony PCR conditions using GoTaq G2 Flexi DNA polymerase:
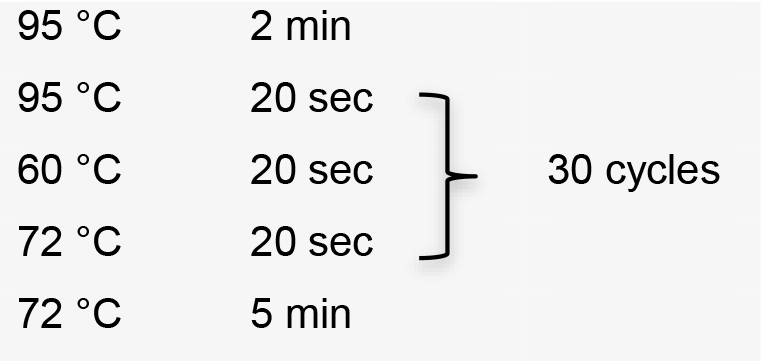
Hold temperature at 10 °C.
Positive clones with sgRNA inserted in the right orientation amplify a band of 190 bp (Figure 3).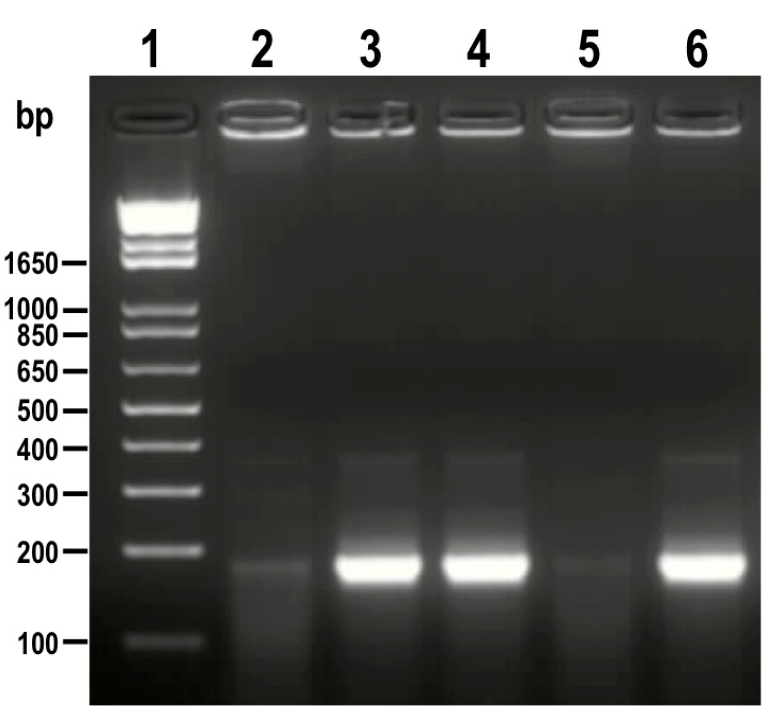
Figure 3. PCR analysis of colonies obtained from cloning of sgRNA into Cas9/pTREX-n vector. A fragment of 190 bp was amplified from positive clones (lanes 3, 4 and 6) while the same band is absent or very weak in negative clones (lanes 2 and 5). 1 kb plus ladder was loaded in lane 1. Molecular weights are indicated on the left side. - Confirm the orientation of sgRNA in positive clones by sequencing using HX1 reverse primer. These clones correspond to construct sgRNA/Cas9/pTREX-n (Figure 4).
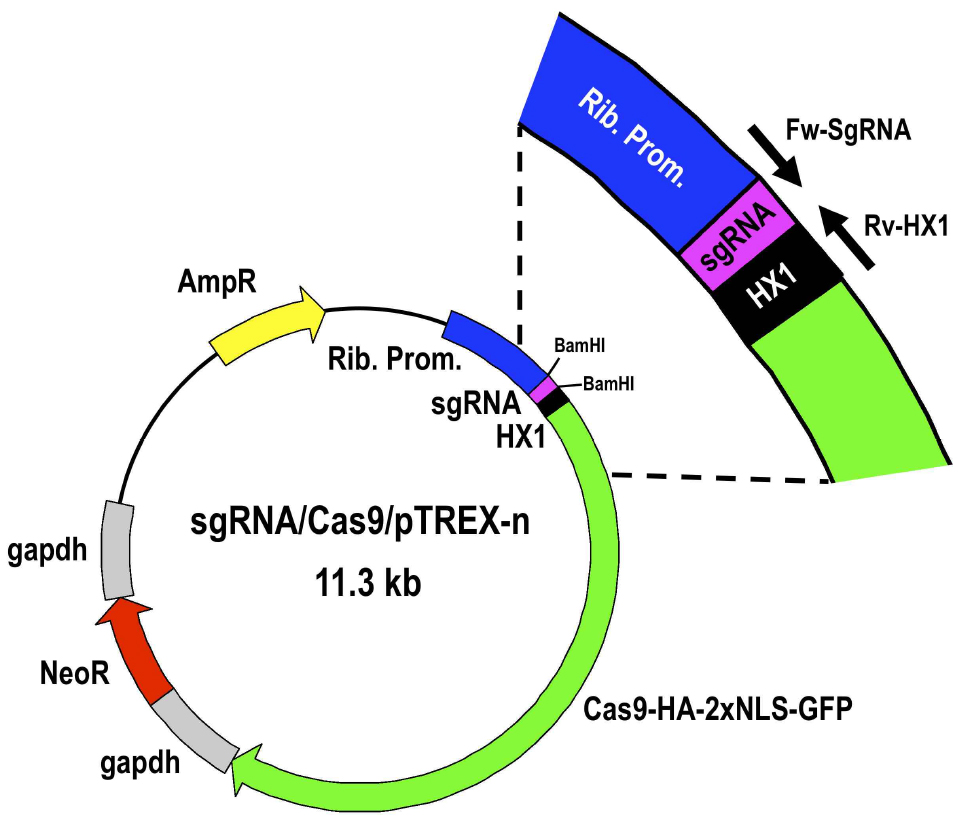
Figure 4. Restriction map of molecular construct sgRNA/Cas9/pTREX-n derived from vector Cas9/pTREX-n by insertion of a specific sgRNA sequence through BamHI restriction site. Rib. Prom, ribosomal promoter; HX1 and gapdh are trans-splicing regions previously described (Vazquez and Levin, 1999); AmpR, ampicillin resistance gene; Neo, neomycin resistance gene; Cas9-HA-2xNLS-GFP is a fusion gene encoding for Cas9 endonuclease as previously described (Lander et al., 2015). - Select one positive clone to isolate enough plasmid DNA by Miniprep for at least 3 transfections (~100 μg).
- Precipitate approximately 100 μg plasmid DNA by adding 1.5 volumes of 100% ethanol and 0.1 volumes of 3 M acetic acid, pH 5.2. Incubate overnight at -20 °C. Centrifuge plasmid DNA at maximum speed for 30 min at 4 °C, remove supernatant and add 1 ml of 70% ethanol. Apply vortex for 15 sec, centrifuge again at maximum speed for 5 min. Remove supernatant, air dry the DNA pellet (do not over-dry to facilitate DNA resuspension). Resuspend DNA in 50 μl ultra-pure water.
- Quantify plasmid DNA using a NanoDrop spectrophotometer.
- Digest 1 μg of insert DNA (sgRNA purified PCR product) and 1 μg of DNA vector (Cas9/pTREX-n) separately with BamHI restriction enzyme (20 U) by incubating overnight at 37 °C.
- DNA donor amplification
- Design ultramers to amplify a DNA cassette for gene tagging by homologous directed repair. First, select a region of 100 bp located right upstream of the stop codon of the target gene (homologous region 1 or HR1). Select a second 100 bp homologous region (HR2) located downstream of the Cas9 cut site on the protospacer. Cas9 cuts 3 nucleotides upstream of the PAM sequence (Figure 1). Determine the reverse complementary sequence of HR2 (HR2rc). Forward ultramer will be as follows: 5’-HR1-GGTACCGGGCCCCCCCTCGAG-3’. Reverse ultramer will be as follows: 5’-HR2rc-TGGCGGCCGCTCTAGAACTAGTGGAT-3’.
- Amplify the DNA donor containing the tag sequence and a marker for antibiotic resistance, plus 100 bp homology arms using pMOHX1Tag4H vector (for 3xHA tagging and hygromycin resistance, Figure 5A) or pMOTag23M vector (for 3xc-Myc tagging and puromycin resistance, Figure 5B) as template (Recipe 3). Perform enough PCR reactions to reach 500-1,000 μl PCR product. PCR conditions using GoTaq G2 Flexi DNA polymerase:
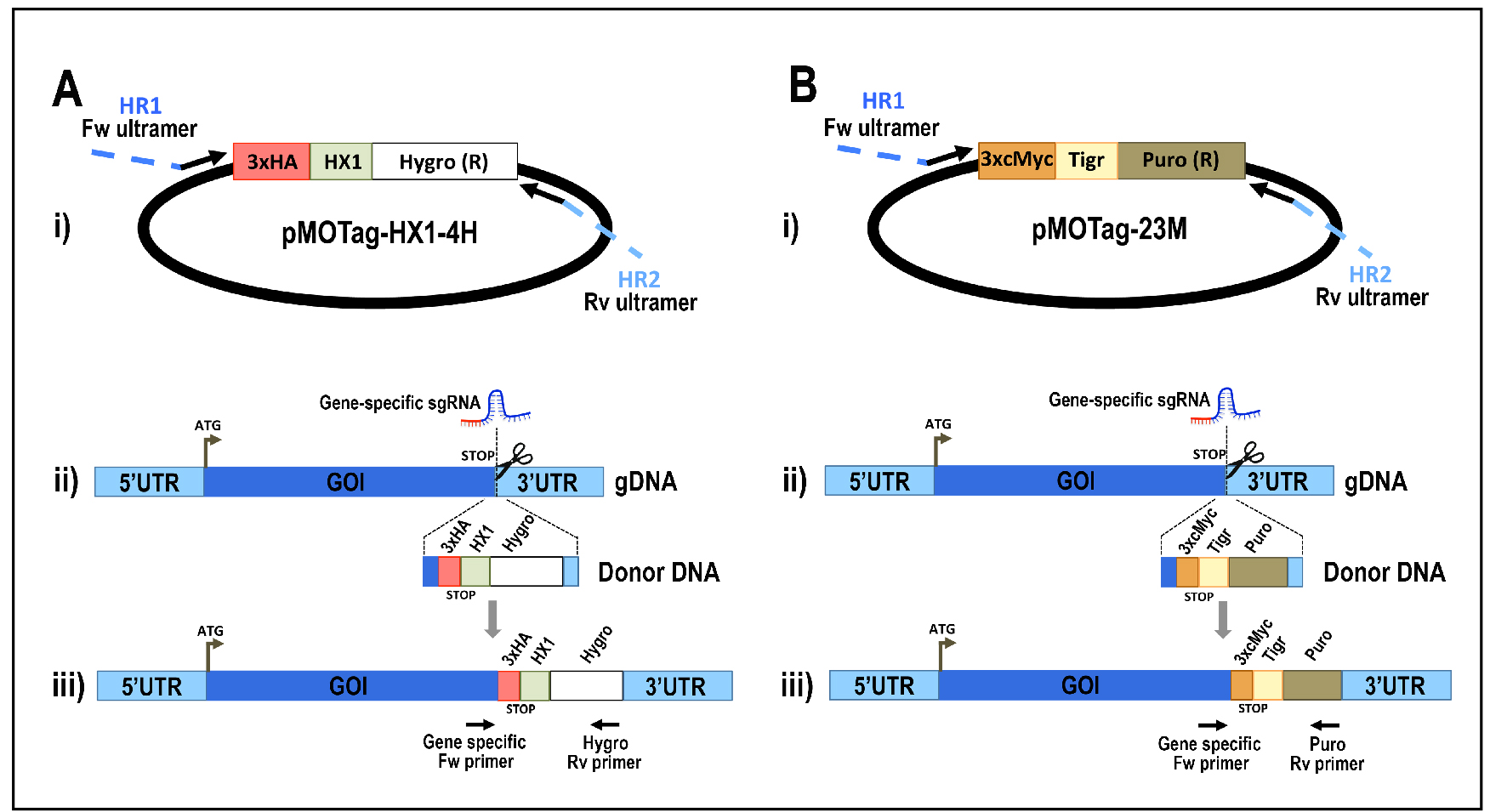
Figure 5. Schematic representation of strategies used to generate endogenous C-terminal tagging in Trypanosoma cruzi. A. i) pMOTag-HX1-4H vector map. The T. cruzi HX1 trans-splicing signal is located between the 3xHA tag sequence and the gene that confers resistance to hygromycin (Hygro R). HR1 Fw and HR2 Rv ultramers indicate oligonucleotides used to amplify DNA donor cassette. The annealing regions for ultramers to pMOTag-HX1-4H and genomic DNA (gDNA) are indicated in black and blue, respectively. ii) A double-stranded gDNA break was produced by Cas9 targeted by the sgRNA both expressed from 3’end-sgRNA/Cas9/pTREX plasmid downstream of the STOP codon of the gene of interest (GOI) in the endogenous locus. Homologous directed repair was induced co-transfecting epimastigotes with the DNA donor cassette, containing homologous regions to the GOI 3’ end (blue) and to the GOI 3’UTR (light blue). iii) Integration of 3xHA and antibiotic resistance gene at 3’end of GOI by homologous recombination. Arrows indicate primers used for checking integration of donor DNA. B. i) pMOTag23M vector map. The 3xc-Myc tag sequence and the puromycin resistance gene (Puro R) are separated by the T. brucei tubulin intergenic region (Tigr). The rest of i), ii) and iii) description is similar to that panel A. Hygro, hygromycin resistance gene; Puro, puromycin resistance gene; UTR, 5’ and 3’ untranslated regions; ATG, start codon. This figure was originally published in Lander et al., 2016b. - Analyze PCR product on agarose gel. PCR product size: 1.5 kb (using pMOTag23M as template) (Figure 6) or 1.6 kb (using pMOHX1Tag4H as template)

Figure 6. DNA donor (1.5 kb) amplified from vector pMOTag23M and visualized on 1% agarose gel (lane 2). 1 kb plus ladder was loaded in lane 1. Molecular weights are indicated on the left side. - Purify 500-1,000 μl PCR product by adding an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) in 1.5 ml tubes, then apply vortex for 1 min, centrifuge at maximum speed for 2 min and carefully transfer the supernatant into a new 1.5 ml tube.
- Precipitate DNA donor by adding 1.5 volumes of 100% ethanol and 0.1 volumes of 3 M acetic acid, pH 5.2. Incubate overnight at -20 °C. Centrifuge DNA donor at maximum speed for 30 min at 4 °C, remove supernatant and add 1 ml of 70% ethanol. Apply vortex for 15 sec, centrifuge again at maximum speed for 5 min. Remove supernatant, air dry the DNA pellet (do not over-dry to facilitate DNA resuspension). Resuspend DNA in 50 μl ultra-pure water.
- Quantify DNA donor using a NanoDrop spectrophotometer.
- Design ultramers to amplify a DNA cassette for gene tagging by homologous directed repair. First, select a region of 100 bp located right upstream of the stop codon of the target gene (homologous region 1 or HR1). Select a second 100 bp homologous region (HR2) located downstream of the Cas9 cut site on the protospacer. Cas9 cuts 3 nucleotides upstream of the PAM sequence (Figure 1). Determine the reverse complementary sequence of HR2 (HR2rc). Forward ultramer will be as follows: 5’-HR1-GGTACCGGGCCCCCCCTCGAG-3’. Reverse ultramer will be as follows: 5’-HR2rc-TGGCGGCCGCTCTAGAACTAGTGGAT-3’.
- Cell culture
- Culture T. cruzi Y strain epimastigotes in liver infusion tryptose (LIT) medium (Recipe 4) containing 10% heat-inactivated FBS and penicillin (100 U/ml)/streptomycin (100 μg/ml) at 28 °C (Bone and Steinert, 1956).
- Determine cell density of T. cruzi cultures by counting cells in a Neubauer chamber. T. cruzi epimastigotes should be manipulated in biological safety cabinets.
- Culture T. cruzi Y strain epimastigotes in liver infusion tryptose (LIT) medium (Recipe 4) containing 10% heat-inactivated FBS and penicillin (100 U/ml)/streptomycin (100 μg/ml) at 28 °C (Bone and Steinert, 1956).
- Cell transfections
- Co-transfect T. cruzi epimastigotes with sgRNA/Cas9/pTREX-n plasmid and DNA donor cassette for gene tagging. Perform transfections in triplicates.
- Grow T. cruzi epimastigotes to a density of 1-2 x 107 cells/ml.
- Wash cells once in PBS pH 7.4 at room temperature.
- Resuspend cells at a density of 1 x 108 cells/ml in ice-cold cytomix (Recipe 5).
- Mix 0.4 ml of cell suspension with 25 μg sgRNA/Cas9/pTREX-n plasmid and 25 μg DNA donor (each DNA in a maximum volume of 20 μl) in ice-cold 0.4 cm electroporation cuvettes. As controls for the electroporation, add 10 μl of ultra-pure water to one cuvette of cells, and add nothing to another cuvette of cells.
- Keep cells in cuvette on ice for 10 min and then electroporate using a Bio-Rad Gene Pulser XcellTM system set at 1.5 kV and 25 μF with 3 pulses, leaving cuvettes for 1 min on ice between pulses. Video 1 shows the standard electroporation procedure described in this protocol.
 Video 1. Electroporation of T. cruzi epimastigotes
Video 1. Electroporation of T. cruzi epimastigotes - Let cells recover in cuvette at room temperature for 15 min.
- Transfer cells to 5 ml LIT media supplemented with 20% heat-inactivated fetal bovine serum and incubate at 28 °C.
- After 24 h, add antibiotics for selection. Antibiotics concentration should be previously optimized for each particular T. cruzi strain. For Y strain, cell lines should be maintained in medium containing 250 μg/ml G418 and 5 μg/ml puromycin (3xc-Myc tagged) or 250 μg/ml G418 and 350 μg/ml hygromycin (3xHA tagged).
- Maintain parasites in LIT medium supplemented with 20% FBS during the selection of resistant cells (usually 4-5 weeks). During this period, replace medium once a week by centrifuging parasites, removing supernatant and adding fresh medium with 20% FBS and antibiotics. In this way parasites are not diluted until reaching a cell density around 1-2 x 107 cells/ml. At this point, FBS concentration can be reduced to 10% in the culture medium and cells can be normally diluted for maintenance.
- Co-transfect T. cruzi epimastigotes with sgRNA/Cas9/pTREX-n plasmid and DNA donor cassette for gene tagging. Perform transfections in triplicates.
Data analysis
- After 4-5 weeks under antibiotic selection, isolate gDNA from double-resistant transfectants as described (Medina-Acosta and Cross, 1993).
- Analyze gDNA by PCR to verify the integration of the DNA donor molecules into the 3’ end of the tagged genes. In this PCR reaction include a gene-specific forward primer annealing upstream of de homology region 1 (HR1) and a reverse primer annealing at the 3’ end of the resistance marker present in the DNA donor (primer Rv_Puro_Ctag_check: 5’-TCAGGCACCGGGCTTGCGGG-3’ for 3xc-Myc tagging and primer Rv_Hygro_Ctag_check: 5’-CTATTCCTTTGCCCTCGGAC-3’ for 3xHA tagging). PCR conditions will vary depending on PCR product size. Figures 7A and 7D show examples of PCR analysis of TcVP1 gene tagged with 3xHA and 3xc-Myc, respectively (Lander et al., 2016b).
- Protein tagging should be confirmed by Western blot and immunofluorescence analysis (IFA) using antibodies anti-HA epitope tag or antibodies anti-c-Myc tag. Figure 7 shows examples of Western blot (Panels B and E) and IFAs (Panels C and F) of TcVP1 protein tagged with 3xHA and 3xc-Myc, respectively (Lander et al., 2016b).
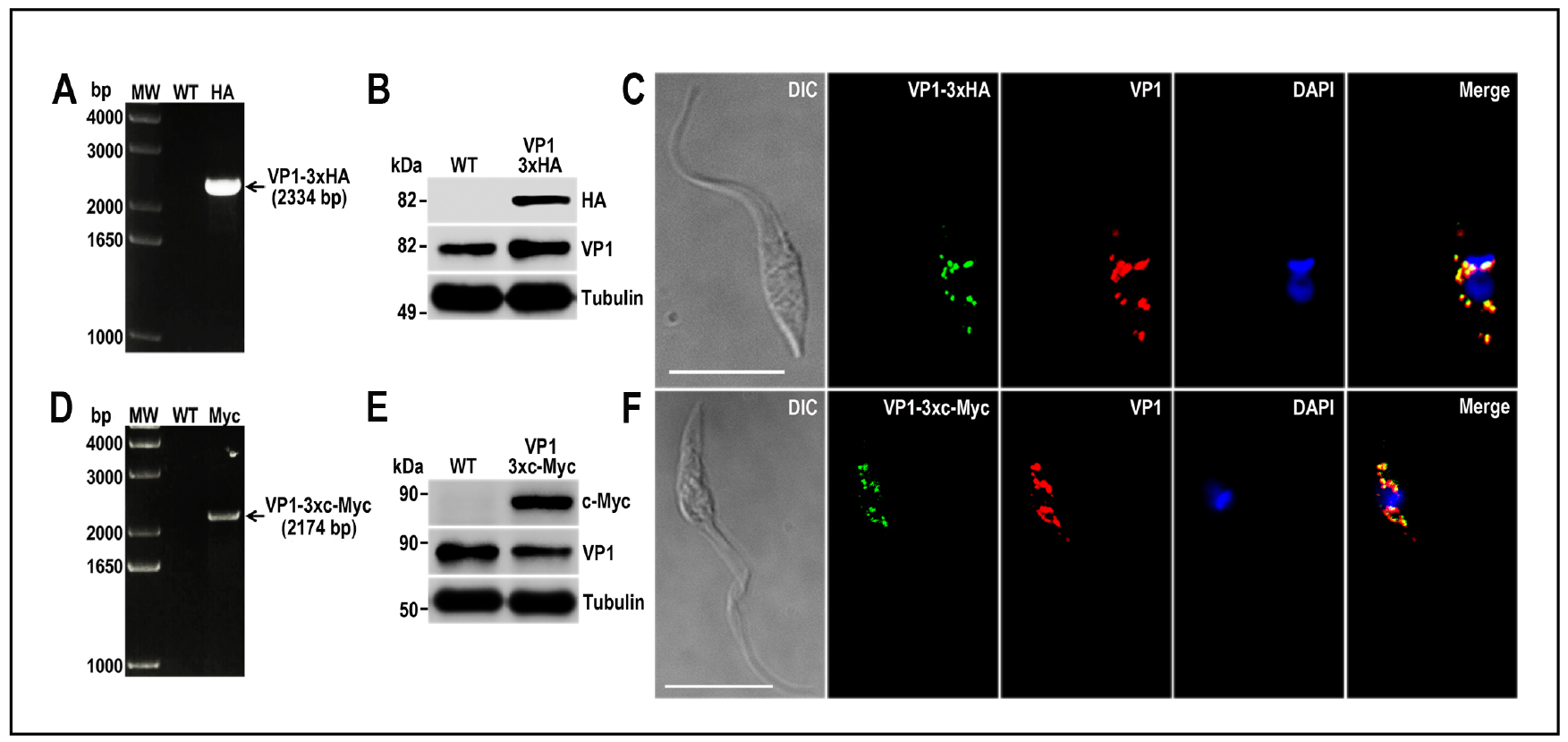
Figure 7. TcVP1 endogenous C-terminal tagging. A. PCR analysis using gDNA isolated from wild type (WT) and TcVP1-3xHA cell lines. A DNA fragment was amplified in 3xHA-tagged epimastigotes (indicated with arrow), while the band is absent in WT. B. Western blot analysis of WT and TcVP1-3xHA cell lines. Anti-HA antibodies detect TcVP1-3xHA (expected size 89 kDa) and anti-TbVP1 antibodies detect endogenous TcVP1 (85 kDa). Anti-α-tubulin antibody was used as a loading control. Antibodies are indicated on the right side of the blots and molecular weights on the left side. C. Fluorescence microscopy of TcVP1-3xHA epimastigotes indicates localization of the endogenous tagged protein to acidocalcisomes. TcVP1-3xHA was detected with monoclonal anti-HA antibodies (green) or with polyclonal anti-TbVtc4 antibodies (red). D. PCR analysis of TcVP1-3xc-Myc epimastigotes. A DNA fragment was amplified in c-Myc-tagged epimastigotes (indicated with arrow), while the band is absent in WT cells. E. Western blot analysis of WT and TcVP1-3xc-Myc cell lines. Anti-c-Myc antibodies detect TcVP1-3xc-Myc (expected size 91 kDa). Anti-TbVP1 antibodies detect endogenous TcVP1 (85 kD). F. Fluorescence microscopy of TcVP1-3xc-Myc epimastigotes indicates localization of the endogenous tagged protein to acidocalcisomes. TcVP1-3xc-Myc was detected with monoclonal anti-c-Myc antibodies (green) or with polyclonal anti-TbVP1 antibodies (red). The merge shows co-localization in yellow. Differential interference contrast (DIC) images are shown on the left panel. Nucleus and kinetoplast were labeled with DAPI (blue). Bars = 10 μm. This figure was originally published in Lander et al., 2016b.
Notes
- Due to the high genome diversity exhibited among different T. cruzi strains, it is critical to use the right genome sequence from TriTrypDB in Procedure A and Procedure D (selection of protospacers and DNA donor amplification) according to the Discrete Typing Unit (DTU) of the T. cruzi strain to be tagged. As an example, for tagging genes of T. cruzi Y strain we use T. cruzi CL Brener Esmeraldo-like (Curated Reference Strain) genomic sequence, for the initial in silico analysis, as the Y strain genomic sequence is not available yet and it belongs to the same DTU as T. cruzi Esmeraldo strain (DTU II). DTUs nomenclature for genetic classification of T. cruzi lineages is available in the literature (Zingales et al., 2009).
- Before proceeding to cell transfection, we strongly recommend to determine the optimal concentration of antibiotics to be used during the selection of tagged parasites. This should be done for the working strain used on each laboratory, even if other research groups have previously reported the antibiotic concentration. We encourage each laboratory to perform this optimization, as antibiotic sensitivity can be very different among strains maintained for long periods under different conditions. The optimal antibiotic concentration should kill the entire population in a T25 flask (~2-3 x 107 cells in 5 ml) in about 3 weeks, when exchanging the medium once a week.
- After confirmation of gene tagging by PCR from genomic DNA, the detection of the tagged protein by Western blot and immunofluorescence analysis will depend on the expression level of the protein in T. cruzi epimastigotes. Further differentiation assays should be performed in order to analyze expression of the tagged protein in other life cycle stages.
Recipes
- sgRNA amplification PCR reaction mix for 1 reaction

- Colony PCR reaction mix for 1 reaction

- Donor DNA PCR reaction mix for 1 reaction

- Hemin stock solution
Prepare 20 mg/ml hemin stock solution by dissolving hemin in 1 N NaOH solution. Store at 4 °C, protected from light - LIT medium (Liver Infusion Tryptose)
68 mM NaCl
5.3 mM KCl
56 mM Na2HPO4
0.2% (w/v) glucose
0.5% (w/v) liver infusion
0.5% (w/v) trypticase
0.002% (w/v) hemin
Note: After dissolving all reagents in MilliQ water, adjust pH to 7.3 and sterilize by autoclave. - Electroporation buffer (cytomix), pH 7.6
120 mM KCl
0.15 mM CaCl2
10 mM K2HPO4
25 mM HEPES
2 mM EDTA
5 mM MgCl2
Note: After dissolving all reagents in MilliQ water, adjust pH to 7.6 and filter sterilize, then storage at 4 °C. - 1x TAE (Tris-acetate-EDTA) buffer, pH 8.3
40 mM Tris base
20 mM acetic acid
1 mM EDTA
Acknowledgments
These experimental procedures have been published (Lander et al., 2016b). We acknowledge Mayara Bertolini for her assistance in recording Video 1. This work was funded by the São Paulo Research Foundation (FAPESP), Brazil (2013/50624-0) and the U.S. National Institutes of Health (grant AI107663). N.L. and M.A.C. are postdoctoral fellows of FAPESP (2014/08995-4 and 2014/13148-9).
References
- Bone, G. J. and Steinert, M. (1956). Isotopes incorporated in the nucleic acids of Trypanosoma mega. Nature 178(4528): 308-309.
- Docampo, R. (2011). Molecular parasitology in the 21st century. Essays Biochem 51:1-13.
- Lander, N., Chiurillo, M. A. and Docampo, R. (2016a). Genome editing by CRISPR/Cas9: a game change in the genetic manipulation of protists. J Eukaryot Microbiol 63(5): 679-690.
- Lander, N., Chiurillo, M. A., Storey, M., Vercesi, A. E. and Docampo, R. (2016b). CRISPR/Cas9-mediated endogenous C-terminal tagging of Trypanosoma cruzi genes reveals the acidocalcisome localization of the inositol 1,4,5-trisphosphate receptor. J Biol Chem 291(49): 25505-25515.
- Lander, N., Li, Z. H., Niyogi, S. and Docampo, R. (2015). CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. MBio 6(4): e01012.
- Medina-Acosta, E. and Cross, G. A. (1993). Rapid isolation of DNA from trypanosomatid protozoa using a simple ‘mini-prep’ procedure. Mol Biochem Parasitol 59(2): 327-329.
- Oberholzer, M., Morand, S., Kunz, S. and Seebeck, T. (2006). A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol 145(1): 117-120.
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A. and Tarleton, R. L. (2014). CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. MBio 6(1): e02097-02014.
- Sidik, S. M., Hackett, C. G., Tran, F., Westwood, N. J. and Lourido, S. (2014). Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One 9(6): e100450.
- Urbina, J. A. and Docampo, R. (2003). Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol 19(11): 495-501.
- Vazquez, M. P. and Levin, M. J. (1999). Functional analysis of the intergenic regions of TcP2β gene loci allowed the construction of an improved Trypanosoma cruzi expression vector. Gene 239(2): 217-225.
- Zingales, B., Andrade, S. G., Briones, M. R., Campbell, D. A., Chiari, E., Fernandes, O., Guhl, F., Lages-Silva, E. A., Macedo, M., Machado, C. R., Miles, M. A., Romanha, A. J., Sturm, N. R., Tibayrenc, M. and Schijman, A. G. (2009). A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz 104(7): 1051-1054.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Lander, N., Chiurillo, M. A., Vercesi, A. E. and Docampo, R. (2017). Endogenous C-terminal Tagging by CRISPR/Cas9 in Trypanosoma cruzi. Bio-protocol 7(10): e2299. DOI: 10.21769/BioProtoc.2299.
- Lander, N., Chiurillo, M. A., Storey, M., Vercesi, A. E. and Docampo, R. (2016b). CRISPR/Cas9-mediated endogenous C-terminal tagging of Trypanosoma cruzi genes reveals the acidocalcisome localization of the inositol 1,4,5-trisphosphate receptor. J Biol Chem 291(49): 25505-25515.
Category
Immunology > Host defense > General
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.