- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9 Editing of the Bacillus subtilis Genome
Published: Vol 7, Iss 8, Apr 20, 2017 DOI: 10.21769/BioProtoc.2272 Views: 21632
Reviewed by: Modesto Redrejo-RodriguezJudd F HultquistVinay Panwar
Original research article
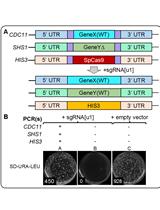
The authors used this protocol in:

Jan 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
A fundamental procedure for most modern biologists is the genetic manipulation of the organism under study. Although many different methods for editing bacterial genomes have been used in laboratories for decades, the adaptation of CRISPR/Cas9 technology to bacterial genetics has allowed researchers to manipulate bacterial genomes with unparalleled facility. CRISPR/Cas9 has allowed for genome edits to be more precise, while also increasing the efficiency of transferring mutations into a variety of genetic backgrounds. As a result, the advantages are realized in tractable organisms and organisms that have been refractory to genetic manipulation. Here, we describe our method for editing the genome of the bacterium Bacillus subtilis. Our method is highly efficient, resulting in precise, markerless mutations. Further, after generating the editing plasmid, the mutation can be quickly introduced into several genetic backgrounds, greatly increasing the speed with which genetic analyses may be performed.
Keywords: Genome editingBackground
Bacillus subtilis is a highly tractable, Gram-positive bacterium. It is amenable to genetic studies, using a variety of vectors to quickly and efficiently introduce mutations by homologous recombination. Although there are many different methods to introduce mutations in B. subtilis, each method has its limitations. A simple and straightforward method to make a mutation in B. subtilis is gene disruption, wherein a plasmid is integrated within a gene of interest (Vagner et al., 1998). The major limitations include: 1) the potential for polar effects by disrupting an operon; 2) introduction and retention of foreign DNA; 3) once an antibiotic resistance cassette is used, the researcher has to use a different cassette if a given mutation is to be studied in the context of other mutations; and 4) the method is limited to targeting an entire gene and cannot yield more precise point mutations. Another method employed in B. subtilis genetic studies is allelic replacement, wherein a gene of interest is replaced with an antibiotic resistance cassette (Guerout-Fleury et al., 1996). Although polar effects should be reduced by simply replacing one gene with another, this method still suffers from several limitations described above. Recently, a gene deletion library was constructed which allows for the removal of the antibiotic resistance cassette (strains are available from the Bacillus genetic stock center). As a result, researchers can use the same method for many mutations because the resistance cassette is removed after each allelic replacement. The method is an improvement, although it is still limited to gene deletions and cannot be used for point mutations. Finally, there are two methods to introduce markerless mutations in B. subtilis including point mutations. One method utilizes the upp gene as a counter-selectable marker (Fabret et al., 2002), and the other uses a plasmid called pMad (Arnaud et al., 2004) or its derivative pMiniMad which allows for mutation integration after removal of the integrating vector (Patrick and Kearns, 2008). Although these methods can introduce precise point mutations, our experience (making four gene deletions and inserting gfp at one genetic locus) using the latter method (Arnaud et al., 2004; Patrick and Kearns, 2008) is that it is quite time consuming with a success rate that is not very high (on average, about 12% success). Although we do not have experience with the upp counter selection method, the authors engineered a GGA→GAC change in the lexA gene and reported the intended change in sequence for three out of four screened isolates with the incorrect isolate yielding multiple mutations in the targeted lexA gene (Fabret et al., 2002). A major drawback, though, is that the method requires deletion of the endogenous upp gene in B. subtilis, which requires that the Δupp strain be used as the new ‘wild-type’ control. Therefore, although the methods described above work, we were in search of a genome editing method with a higher efficiency that also required less time at the bench. These criteria prompted us to adapt a CRISPR/Cas9 genome editing system (Jiang et al., 2013) to B. subtilis (Burby and Simmons, 2017). CRISPR/Cas9 can be used to introduce a variety of mutations including gene deletions, fusions, and even point mutations (Sternberg and Doudna, 2015). Further, by constructing the editing system on a single broad host-range plasmid with a temperature sensitive origin of replication, all vector DNA introduced during the procedure can easily be removed. Success rates have proven to be much higher for point mutations and small, gene-sized deletions (often over 80% success, but 100% success is not atypical), reducing the number of isolates that need to be screened. Although this method solves many of the limitations of the contemporary methods, our CRISPR/Cas9 genome editing system is limited by the requirement of a proto-spacer adjacent motif or PAM sequence (NGG in our system), and the requirement of two cloning steps. Nonetheless, the ability to make a variety of markerless mutations, coupled with the rapidity with which the mutations can be transferred to different genetic backgrounds still provides a significant improvement over current methods for genome editing in B. subtilis. The system we have developed may also be applicable to other Gram-positive bacteria with little or no manipulation of the DNA reagents described herein.
Materials and Reagents
- Pipette tips:
with boxes (USA Scientific, catalog numbers: 1161-3800 ; 1161-1800 and 1161-1820 )
for refills (USA Scientific, catalog numbers: 1161-3700 ; 1161-1700 and 1161-1720 ) - Microfuge tubes (Fisher Scientific, catalog number: 02-681-320 )
- PCR tubes (Fisher Scientific, catalog number: 14-230-225 )
- Wooden sticks for colony purification/bacteria re-streaking (Ted Pella, catalog number: 1282 )
- Round bottom culture tube (Fisher Scientific, catalog number: 14-956-6D )
- Cryo-vials (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 368632 )
- Cryo-vial caps (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 375930 )
- Silica spin columns for gel extraction and plasmid mini-prep (Epoch Life Science, catalog number: 1920250 )
- Petri dishes (Fisher Scientific, catalog number: FB0875712 )
- pPB41 plasmid (plasmid and sequence available from the Bacillus Genetic Stock Center upon request; http://www.bgsc.org)
- pPB105 plasmid (plasmid and sequence available from the Bacillus Genetic Stock Center upon request; http://www.bgsc.org)
- TOP10 Escherichia coli competent cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: C404010 )
- MC1061 Escherichia coli competent cells (Strain number PEB336, available upon request)
- BsaI-HF with accompanying 10x Cutsmart buffer (New England Biolabs, catalog number: R3535L )
- Calf intestinal alkaline phosphatase (CIP) (New England Biolabs, catalog number: M0290L )
- Isopropanol (Fisher Scientific, catalog number: A451-4 )
- Ultra-pure distilled H2O (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977-15 )
- T4 DNA ligase with accompanying 10x T4 ligase buffer (New England Biolabs, catalog number: M0202L )
- T4 polynucleotide kinase (New England Biolabs, catalog number: M0201S )
- Q5 DNA polymerase (New England Biolabs, catalog number: M0491L )
- PCR primers (synthesized by Integrated DNA Technologies)
- Primers to amplify pPB41
- oPEB217: 5’-GAACCTCATTACGAATTCAGCATGC
- oPEB218: 5’-GAATGGCGATTTTCGTTCGTGAATAC
- Primers to amplify CRISPR/Cas9
- oPEB232: 5’-GCTGTAGGCATAGGCTTGGTTATG
- oPEB234:
5’-GTATTCACGAACGAAAATCGCCATTCCTAGCAGCACGCCATAGTGACTG - Primer to sequence CRISPR insert
oPEB253: 5’-GAAGGGTAGTCCAGAAGATAACGA - Primer to sequence 5’ side of editing template
oPEB227: 5’-CCGTCAATTGTCTGATTCGTTA - Example upstream editing template primers
- oPEB237:
5’-GCATGCTGAATTCGTAATGAGGTTCAAAACGGCAGAGTATACAGAGGAG - oPEB238: 5’-CCGGTTCCTTTTCCAGCGATGATTGACACTCTTGGATATCCG
- Example downstream editing template primers
- oPEB239: 5’-AAGAGTGTCAATCATCGCTGGAAAAGGAACCGGCGCTTTAAG
- oPEB240: 5’-GCATAACCAAGCCTATGCCTACAGCtaggaagaagaatcatttcgaagc
- Example genotyping primer for H743A mutation in mutS2
- oPEB262: 5’- GGATATCCAAGAGTGTCAATCATCGCT
- Glycerol (Fisher Scientific, catalog number: BP229-4 )
- Tris base (Fisher Scientific, catalog number: BP152-5 )
- Glacial acetic acid (Fisher Scientific, catalog number: A38-212 )
- EDTA (Fisher Scientific, catalog number: BP120-1 )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: S271-10 )
- Tryptone (BD, BactoTM, catalog number: 211699 )
- Yeast extract (BD, BactoTM, catalog number: 212720 )
- Ampicillin (Fisher Scientific, catalog number: BP1760-25 )
- Spectinomycin (MP Biomedicals, catalog number: 0 215206725 )
- Chloramphenicol (Fisher Scientific, catalog number: BP904-100 )
- Agar (Acros Organics, catalog number: 400400050 )
- Agarose (Fisher Scientific, catalog number: BP1356-500 )
- Ethidium bromide (Sigma-Aldrich, catalog number: E8751-25G )
- Guanidine thiocyanate (Fisher Scientific, catalog number: BP221-1 )
- Guanidine HCl (Fisher Scientific, catalog number: BP178-1 )
- Ethanol (Decon Labs, catalog number: 2701 )
- Hydrochloric acid (HCl) (Fisher Scientific, catalog number: A144-212 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266-100G )
- 100 mM dNTP set (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10297018 )
- Dithiothreitol (DTT) (Fisher Scientific, catalog number: BP172-25 )
- β-nicotinamide adenine dinucleotide (NAD+) (Acros Organics, catalog number: 124530010 )
- T5 exonuclease (New England Biolabs, catalog number: M0363S )
- Phusion DNA polymerase (New England Biolabs, catalog number: M0530L )
- Taq DNA ligase (New England Biolabs, catalog number: M0208L )
- Magnesium sulfate (MgSO4) (Fisher Scientific, catalog number: M65-500 )
- Potassium phosphate dibasic (K2HPO4) (Fisher, catalog number: BP363-1 )
- Potassium phosphate monobasic (KH2PO4) (Fisher Scientific, catalog number: BP362-1 )
- Trisodium citrate dihydrate (C6H5Na3O7·2H2O) (Fisher Scientific, catalog number: BP327-1 )
- Glucose (Sigma-Aldrich, catalog number: G8270-1KG )
- Tryptophan (Fisher Scientific, catalog number: BP395-100 )
- Phenylalanine (Fisher Scientific, catalog number: BP391-100 )
- Potassium hydroxide (KOH) (Fisher Scientific, catalog number: P250-500 )
- Ferric ammonium citrate (Sigma-Aldrich, catalog number: F5879 )
- Potassium aspartate (Sigma-Aldrich, catalog number: A6558 )
- PEG-8000 (Dot Scientific, catalog number: DSP48080-500 )
- 50x TAE (see Recipes)
- 1x TAE (see Recipes)
- LB media (see Recipes)
- LB agar plates (see Recipes)
- 100 mg/ml ampicillin stock (see Recipes)
- 100 mg/ml spectinomycin stock (see Recipes)
- 5 mg/ml chloramphenicol stock (see Recipes)
- 70% (v/v) ethanol (see Recipes)
- 1% agarose gel (see Recipes)
- QG buffer (see Recipes)
- PB buffer (see Recipes)
- Tris HCl, pH 7.5 (see Recipes)
- PE buffer (see Recipes)
- 10x annealing buffer (see Recipes)
- 5x isothermal reaction buffer (Gibson, 2011) (see Recipes)
- 2x Gibson master mix (Gibson, 2011) (see Recipes)
- 1 M MgSO4 (see Recipes)
- LM media (LB media + 3 mM MgSO4) (see Recipes)
- 10x PC buffer (see Recipes)
- MD media (see Recipes)
- 0.1 N KOH (see Recipes)
- 100 mg/ml tryptophan stock (see Recipes)
- 100 mg/ml phenylalanine stock (see Recipes)
- 2.2 mg/ml ferric ammonium citrate stock (see Recipes)
- 100 mg/ml potassium aspartate stock (see Recipes)
Equipment
- Incubator (Napco, model: 320 )
- Dry heat block (Fisher Scientific, model: Fisher ScientificTM IsotempTM Digital Dry Baths/Block Heaters , catalog number: 11-718-2)
- Pipettes (Eppendorf, catalog number: 022575442 )
- Centrifuge (Eppendorf, model: 5424 )
- Thermocycler (Eppendorf, model: 6325 )
- Electrophoresis apparatus (Bio-Rad Laboratories, model: Mini-Sub® Cell GT Horizontal Electrophoresis System , catalog number: 1704406)
- Power source for electrophoresis (Bio-Rad Laboratories, model: PowerPacTM Basic Power Supply , catalog number: 1645050)
- Roller drum (Eppendorf, New Brunswick, model: TC-7 )
- Milli-Q H2O dispenser (Thermo Fisher Scientific, Thermo ScientificTM, model: BarnsteadTM GenPureTM Pro , catalog number: 50131950)
- Microwave (Panasonic, catalog number: NNS954WFR )
- Autoclave
Software
- Online tool Primer3 (Koressaar and Remm, 2007; Untergasser et al., 2012)
- Oligocalc (Kibbe, 2007)
Procedure
- Construction of plasmid containing spacer used to target the B. subtilis genome
- Prepare pPB41 digested with the restriction endonuclease BsaI (Figure 1)
Assemble 50 μl restriction digest reaction: 1x Cutsmart buffer (NEB), 2 μl BsaI-HF (20,000 U/ml; NEB), and 5 μg pPB41. Incubate reaction at 37 °C in an incubator for 3 to 6 h. Add 1 μl of calf intestinal phosphatase (CIP; 10,000 U/ml; NEB), and incubate at 37 °C for 1 h. Perform 1% agarose gel electrophoresis in 1x TAE buffer (see Recipes), followed by gel extraction of the digested plasmid.
Notes:- The plasmid pPB41 is typically used, but we also generated a derivative, pPB105, wherein the spectinomycin resistance cassette was replaced with a chloramphenicol resistance cassette (see Figure 1). All steps of the procedure are identical for pPB105, except for E. coli clones are selected using LB agar plates containing 100 μg/ml ampicillin or 20 μg/ml chloramphenicol (chloramphenicol selection works but colonies grow slower), and B. subtilis transformants are selected using LB agar plates containing 5 μg/ml chloramphenicol.
- Gel extractions are performed by excising the appropriate DNA band from the agarose gel from two 50 μl PCR reactions and placing in a microfuge tube. The gel slice is dissolved by adding 350 μl QG buffer (see Recipes) and incubating at 65 °C in a heat block. After the gel slice is fully dissolved, 200 μl of isopropanol is added and mixed by pipetting. The entire gel extraction is loaded onto a silica spin column. The flow-through is collected via centrifugation (12,000 x g for 1 min at room temperature) and discarded. The column is washed and the flow-through discarded first with 500 μl PB buffer (see Recipes), then with 750 μl PE buffer (see Recipes). The column is dried by centrifugation as above. The DNA is eluted from the column with 75 μl ultra-pure H2O via centrifugation as above.
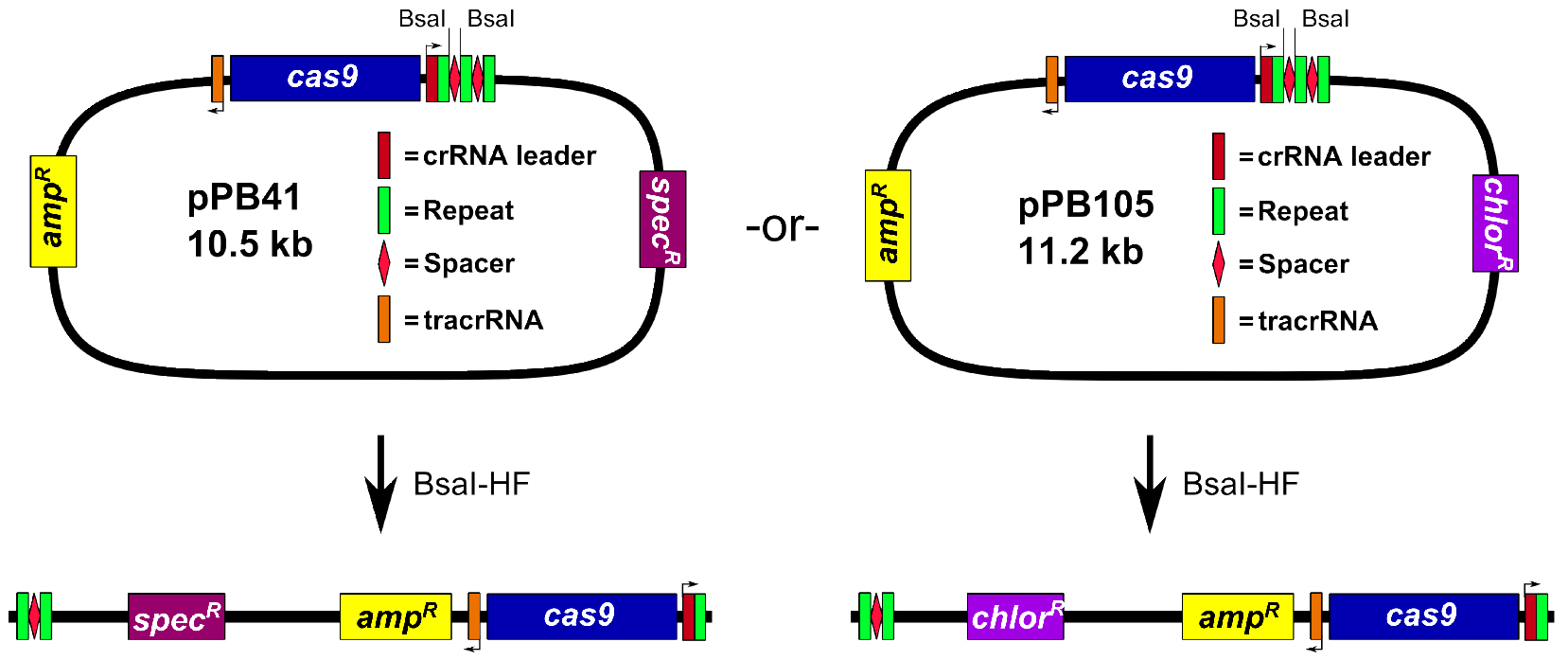
Figure 1. Digestion of pPB41 or pPB105 with BsaI-HF
- The plasmid pPB41 is typically used, but we also generated a derivative, pPB105, wherein the spectinomycin resistance cassette was replaced with a chloramphenicol resistance cassette (see Figure 1). All steps of the procedure are identical for pPB105, except for E. coli clones are selected using LB agar plates containing 100 μg/ml ampicillin or 20 μg/ml chloramphenicol (chloramphenicol selection works but colonies grow slower), and B. subtilis transformants are selected using LB agar plates containing 5 μg/ml chloramphenicol.
- Prepare phosphorylated proto-spacer (Figure 2)
- The proto-spacer is designed by searching the B. subtilis genome for a proto-spacer adjacent motif (PAM) site, consisting of the nucleotide sequence 5’-NGG-3’ (where N stands for any nucleotide), at the location that the edit is to be made (Figure 2A). The thirty nucleotides 5’ of the PAM site are used for the proto-spacer (Figure 2A). Oligonucleotides for the proto-spacer are ordered from Integrated DNA Technologies (IDT) as shown in Figure 2B. The two oligonucleotides are reverse complementary to each other with the exception of the overhangs (Figure 2B). Prepare oligonucleotide stocks at a concentration of 100 μM using ultra-pure distilled H2O (Invitrogen Life Technologies).
- Assemble annealing reaction: 1x annealing buffer (see Recipes), 10 μM oligonucleotide 1, 10 μM oligonucleotide 2. Incubate reaction at 100 °C in a heat block for 5 min. The reaction is transferred to a beaker of water, pre-heated to 100 °C. The entire beaker is placed at room temperature and allowed to cool slowly to room temperature.
- Prepare phosphorylation reaction: 1x T4 DNA ligase buffer (NEB), 1 μl T4 polynucleotide kinase (PNK; 10,000 U/ml; NEB), 1 μM annealed oligonucleotides from above. Incubate at 37 °C in an incubator for 30 min, and then heat inactivate T4 PNK at 65 °C in a heat block for 20 min. Store annealed and phosphorylated oligonucleotides at -20 °C until ready to use in ligation reaction.
Notes: - Although we typically phosphorylate the proto-spacer using T4 PNK, it is also possible to order the oligonucleotides with a 5’-phosphate modification from IDT.
- Given that the T4 PNK inactivation is carried out at 65 °C, it is possible that the proto-spacer could denature. Although we have not had a need to change the order of the protocol (and we have not tried), T4 PNK is active on both single stranded and double stranded DNA, and therefore, we see no theoretical concern with reversing the order, and phosphorylating prior to annealing.
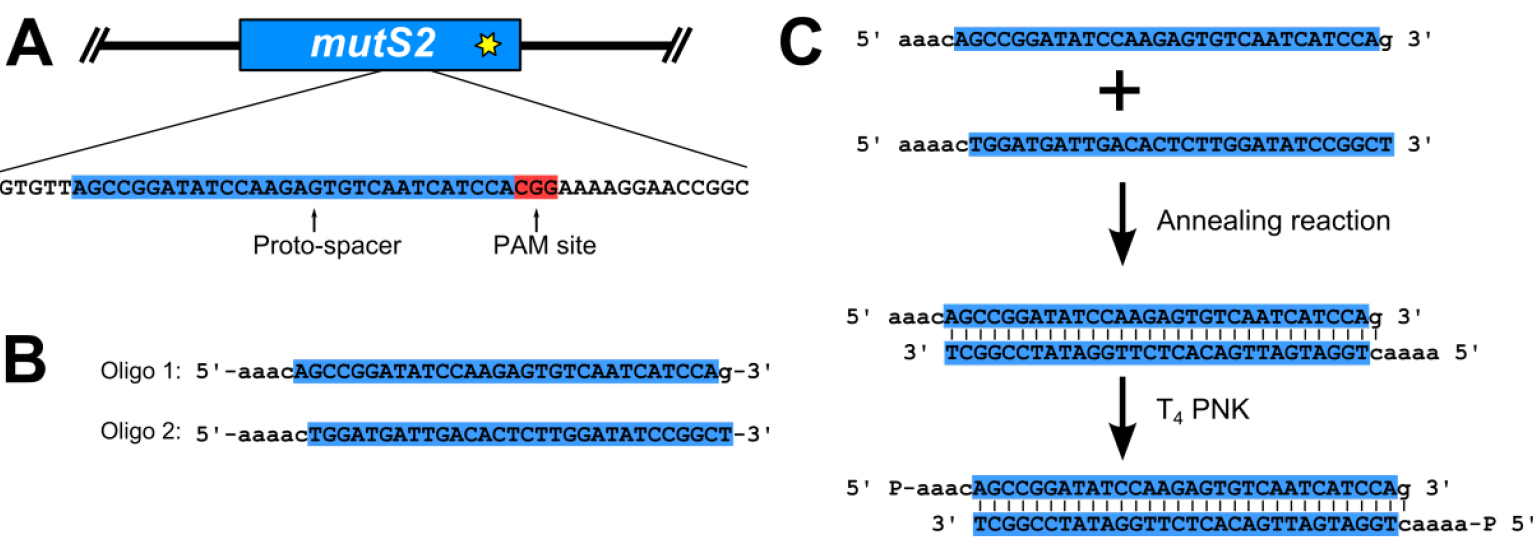
Figure 2. Proto-spacer design and preparation. A. A proto-spacer (highlighted blue and capital letters) and proto-spacer adjacent motif (PAM site; highlighted red); B. Oligonucleotides with proper overhangs (lower case letters) necessary to ligate into pPB41 (Jiang et al., 2013; Burby and Simmons, 2017); C. Annealing and phosphorylation reaction to prepare a dsDNA proto-spacer for ligation into pPB41. - Ligate proto-spacer and pPB41 and isolate new plasmid (Figure 3)
Assemble 20 μl ligation reaction: 1x T4 DNA ligase buffer (NEB), 40 to 100 ng pPB41 (digested with BsaI-HF and CIP in previous step), 25 nM proto-spacer (annealed and phosphorylated in previous step), 1 μl T4 DNA ligase (400,000 U/ml NEB). Let reaction stand at room temperature for 2-3 h. Use 10 μl of ligation reaction to transform 100 μl of chemically competent TOP10 Escherichia coli. Clones can be selected using LB agar plates (see Recipes) containing 100 μg/ml ampicillin or 100 μg/ml spectinomycin. Clones are verified via Sanger sequencing using oPEB253.
Note: The first cloning step, wherein the proto-spacer is ligated into pPB41, does not require a certain E. coli strain. We use TOP10 cells as our general cloning strain.
Figure 3. Ligation of phosphorylated proto-spacer and pPB41 to generate a ‘targeting plasmid’
- Prepare pPB41 digested with the restriction endonuclease BsaI (Figure 1)
- Construction of editing plasmid
- PCR amplify pPB41 to linearize (Figure 4A)
Amplify pPB41 via PCR using Q5 DNA polymerase (NEB) and primers oPEB217 and oPEB218. Perform 1% agarose gel electrophoresis in 1x TAE buffer (see Recipes) followed by gel extraction. PCR product is stored at -20 °C until used.
Notes: - The template used for this step could be either pPB41 or the plasmid containing the spacer generated in step B1.
- The PCR program used to linearize pPB41 is:
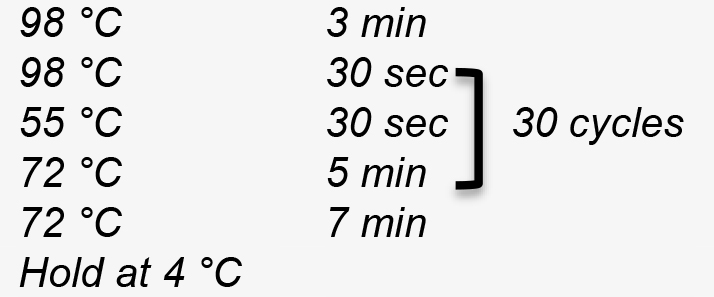
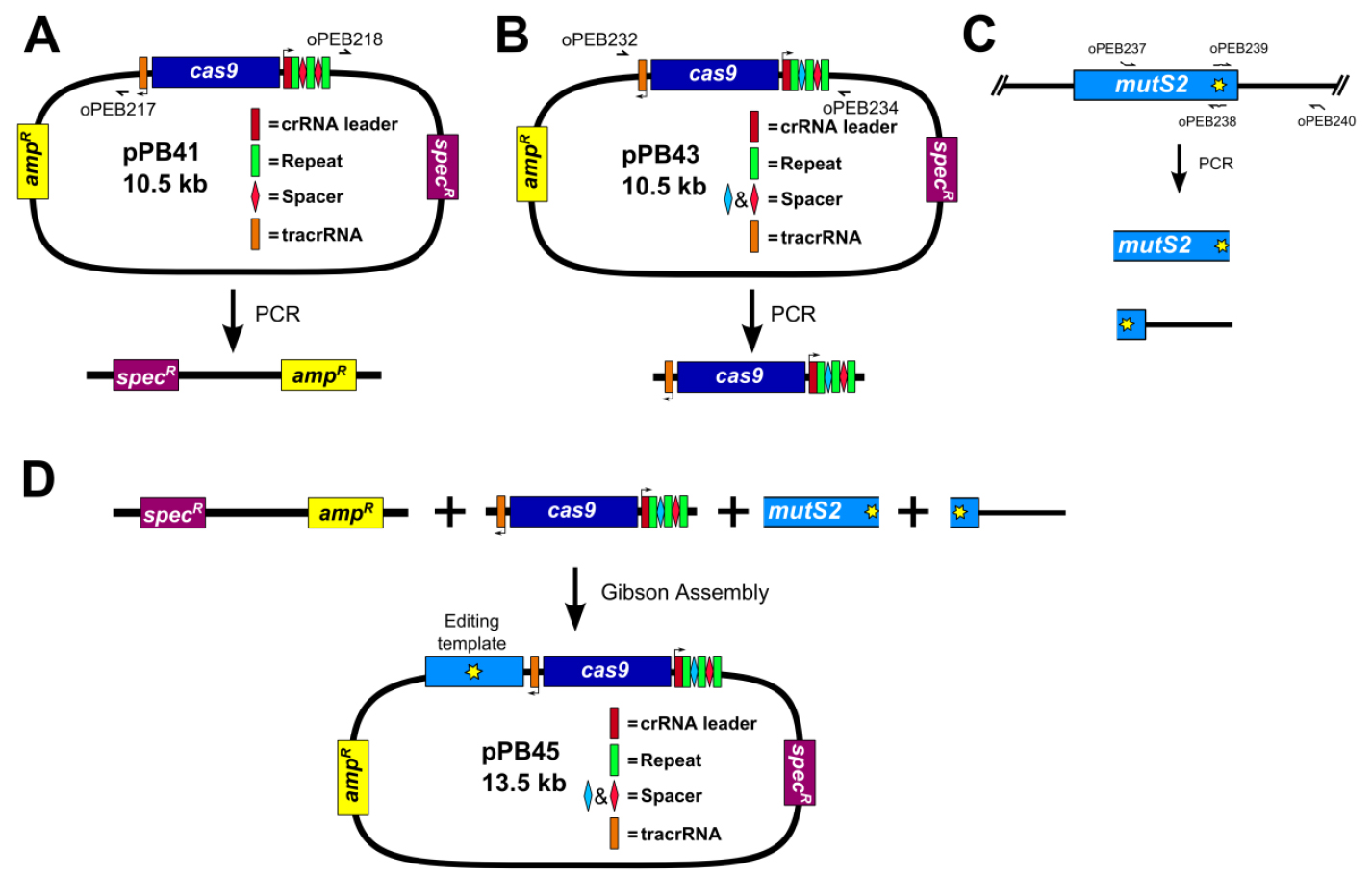
Figure 4. Construction of editing plasmid. A. Schematic representation of pPB41 PCR amplicon; B. Schematic representation of CRISPR/Cas9 PCR amplicon; C. Schematic representation of upstream and downstream portions of editing template PCR amplicons; D. Gibson Assembly of editing plasmid using PCR products from A-C. - PCR amplify CRISPR/Cas9 using plasmid generated in step B1 (Figure 4B)
Amplify plasmid generated in step B1 (pPB43 in the example) via PCR using Q5 DNA polymerase (NEB) and primers oPEB232 and oPEB234. Perform 1% agarose gel electrophoresis in 1x TAE buffer (see Recipes) followed by gel extraction. PCR product is stored at -20 °C until used.
Note: The PCR program used to amplify CRISPR/Cas9 is the same used in step B1 above. - PCR amplify editing template (Figure 4C)
Amplify upstream and downstream portions of editing template separately via PCR using Q5 DNA polymerase (NEB) (primers used in the example are oPEB237/oPEB238 and oPEB239/oPEB240 for the upstream and downstream portions, respectively). Primers are designed using the online tool Primer3 (Koressaar and Remm, 2007; Untergasser et al., 2012) and Oligocalc (Kibbe, 2007). The primers specific to the editing template are combined with the overhangs for pPB41 (upstream forward primer starts 5’-GCATGCTGAATTCGTAATGAGGTTC, followed by the editing template specific sequence; downstream reverse primer starts 5’-GCATAACCAAGCCTATGCCTACAGC, followed by editing template specific sequence), which will allow the editing template to be incorporated into the editing plasmid. The editing template is designed to be approximately 2 kilobases, with the upstream and downstream portions consisting of about 1 kb each. The critical aspect of editing template design is ensuring that the guided Cas9 can no longer target the genome after the desired mutation is introduced. For deletions, the entire sequence of the spacer and the PAM site can be removed. For point mutations, it is more complicated. In Jiang et al. (2013), it was found that mutating the PAM site or the first few bases of the spacer is optimal (Jiang et al., 2013). The overlap at the center of the editing template should be 25 bases on each portion of the template (the result is that there are 50 bp that overlap in total). After PCR, perform 1% agarose gel electrophoresis in 1x TAE buffer (see Recipes) followed by gel extraction. PCR products are stored at -20 °C until used.
Notes: - Editing template primers are designed using the online tool Primer3 (Koressaar and Remm, 2007; Untergasser et al., 2012). Approximately 250 bp is selected at least 750 bp from the center of the editing template and a primer list is generated using default parameters with the following exceptions: 1) primer size used is min: 22, opt: 24, max: 28; 2) primer GC% min: 35; 3) Max Self Complementarity: 6.0; and 4) Max 3’ Self Complementarity: 2.0. A primer is then selected and cross examined using a second online tool, Oligocalc (Kibbe, 2007). The purpose is to ensure that potential hairpin formation, 3’ complementarity, and self-annealing are all undetectable using this program.
- The editing template used in the example in the figures is 3 kb (about 1.5 kb each for upstream and downstream portions of the editing template), but we routinely use 2 kb. We also present four different gene deletions made using editing templates of about 2 kb.
- The PCR program used to amplify each portion of the editing template is:

- Construct editing plasmid using Gibson Assembly (Gibson, 2011) (Figure 4D)
Assemble 10 μl Gibson Assembly reaction: 1x Gibson master mix (see Recipes), 40-100 ng of linearized pPB41 from above, 40-100 ng of CRISPR/Cas9 PCR product containing spacer from above, and 20-40 ng of each portion of the editing template. Incubate reaction at 50 °C in a thermocycler for 90 min. Use 5 μl of Gibson Assembly reaction to transform 80 μl chemically competent E. coli strain MC1061. Clones can be selected as in step A3. Clones are sequenced using primers oPEB253, oPEB227, and editing template specific primers.
Notes:- It is absolutely critical to use a cloning strain that is recA+, because transformation of naturally competent B. subtilis is much more efficient using plasmids that are multimeric. The E. coli strain we use for plasmid propagation is MC1061. We have also encountered difficulties in obtaining certain editing plasmids in MC1061 for reasons that are not clear. In these instances, we scaled up the Gibson Assembly reaction to a total volume of 20 μl and we used 20 μl to transform B. subtilis as in Procedure C. The efficiency of this procedure was lower, with some spectinomycin sensitive isolates retaining the wild-type genotype, so typically at least twelve isolates were screened.
- The editing template is sequenced using oPEB227, which sequences from the 5’ end of pPB41. Additional primers are designed within the editing template to verify the remaining sequence.
- It is absolutely critical to use a cloning strain that is recA+, because transformation of naturally competent B. subtilis is much more efficient using plasmids that are multimeric. The E. coli strain we use for plasmid propagation is MC1061. We have also encountered difficulties in obtaining certain editing plasmids in MC1061 for reasons that are not clear. In these instances, we scaled up the Gibson Assembly reaction to a total volume of 20 μl and we used 20 μl to transform B. subtilis as in Procedure C. The efficiency of this procedure was lower, with some spectinomycin sensitive isolates retaining the wild-type genotype, so typically at least twelve isolates were screened.
- PCR amplify pPB41 to linearize (Figure 4A)
- Editing genome of B. subtilis
- Transform B. subtilis with editing plasmid obtained from previous step
- Prepare naturally competent B. subtilis cultures (Burby and Simmons, 2017) (Figure 5A).
Streak out strain from frozen 20% glycerol stock on LB agar plate for single colonies using a wooden stick and incubate plate at 30 or 37 °C overnight in an incubator. - In the morning, inoculate 2 ml LM media (see Recipes) in a 14 ml disposable, round-bottom culture tube. Incubate culture at 37 °C on a roller drum in an incubator until the OD600 is between 0.8 and 1.5 (approximately 2 h growth).
- Transfer 20 μl of LM culture to 0.5 ml pre-warmed MD media (see Recipes). Incubate MD culture at 37 °C for 4 to 6 h on a roller drum.
- Add editing plasmid DNA (approximately 200 to 600 ng) and incubate at 37 °C on roller drum for 60 to 90 min. Transformants are selected by plating 200 μl on LB agar plates containing 100 μg/ml spectinomycin and incubated at 30 °C overnight.
Notes:- We have found 5 h in MD media to work very well, and is used most often, but adding DNA up to an hour early or an hour late is also successful.
- Plating 90 min after DNA addition works very well, but plating at 60 min will also yield transformants.
- We have found 5 h in MD media to work very well, and is used most often, but adding DNA up to an hour early or an hour late is also successful.
- Several isolates (6-12) are established by colony purifying transformants (Figure 5B)
- Colony purifying refers to restreaking isolated colonies three to four consecutive times for single colonies.
- Single colonies from preceding step are re-streaked on LB agar plates for single colonies containing 100 μg/ml spectinomycin and incubated at 30 °C overnight in an incubator.
- Colony purifying refers to restreaking isolated colonies three to four consecutive times for single colonies.
- Cure isolates of plasmid (Figure 5C)
Single colonies from preceding step are re-struck on LB agar plates for single colonies and incubated at 42-45 °C overnight.
Note: We have an incubator set to 45 °C, but we have used 42 °C successfully.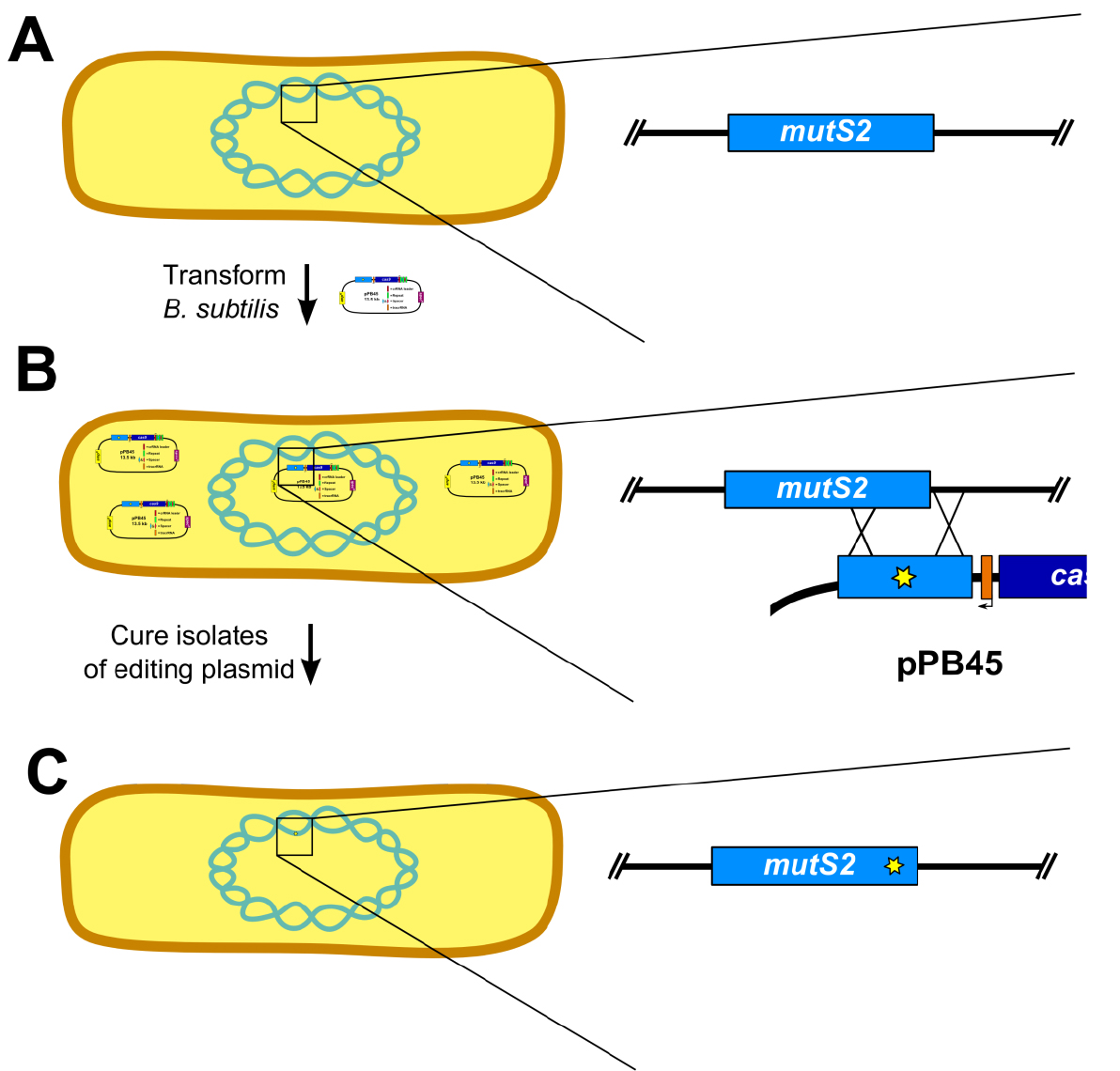
Figure 5. Outline of genome editing procedure in B. subtilis (Adapted from Figure S2 [Burby and Simmons, 2017]). A. The first step is to transform B. subtilis with the editing plasmid. Inset shows wild-type mutS2 locus. B. After establishing isolates using colony purification, the isolates are cured of the editing plasmid. Inset depicts homology directed repair using the editing template on the plasmid. C. The final step is to verify loss of the editing plasmid and genotype isolates to verify the mutation was successfully introduced. Inset shows edited mutS2 locus. - Screen isolates for loss of plasmid
Single colonies from preceding step are re-streaked onto LB agar plates containing 100 μg/ml spectinomycin, and then onto LB agar plates for single colonies. Plates are incubated at 45 °C overnight in an incubator. If the isolates lost the plasmid, there would be no growth on the LB agar plates with spectinomycin. - Screen for intended mutation (Figure 6)
The isolates that were unable to grow on plates containing spectinomycin are further screened for the presence of the desired mutation. In the case of a point mutation, a primer can be designed to anneal at the point mutation (Figure 6A), or the genetic locus may be amplified using primers outside the editing template as done for a deletion (see below), followed by Sanger sequencing to verify the point mutation is present and no other mutations were introduced. In the case of a gene deletion, PCR primers that anneal outside of the recombination template within the genomic DNA are used to PCR amplify the genetic locus (Figure 6B). The migration of the PCR product will be faster relative to the product from wild-type locus.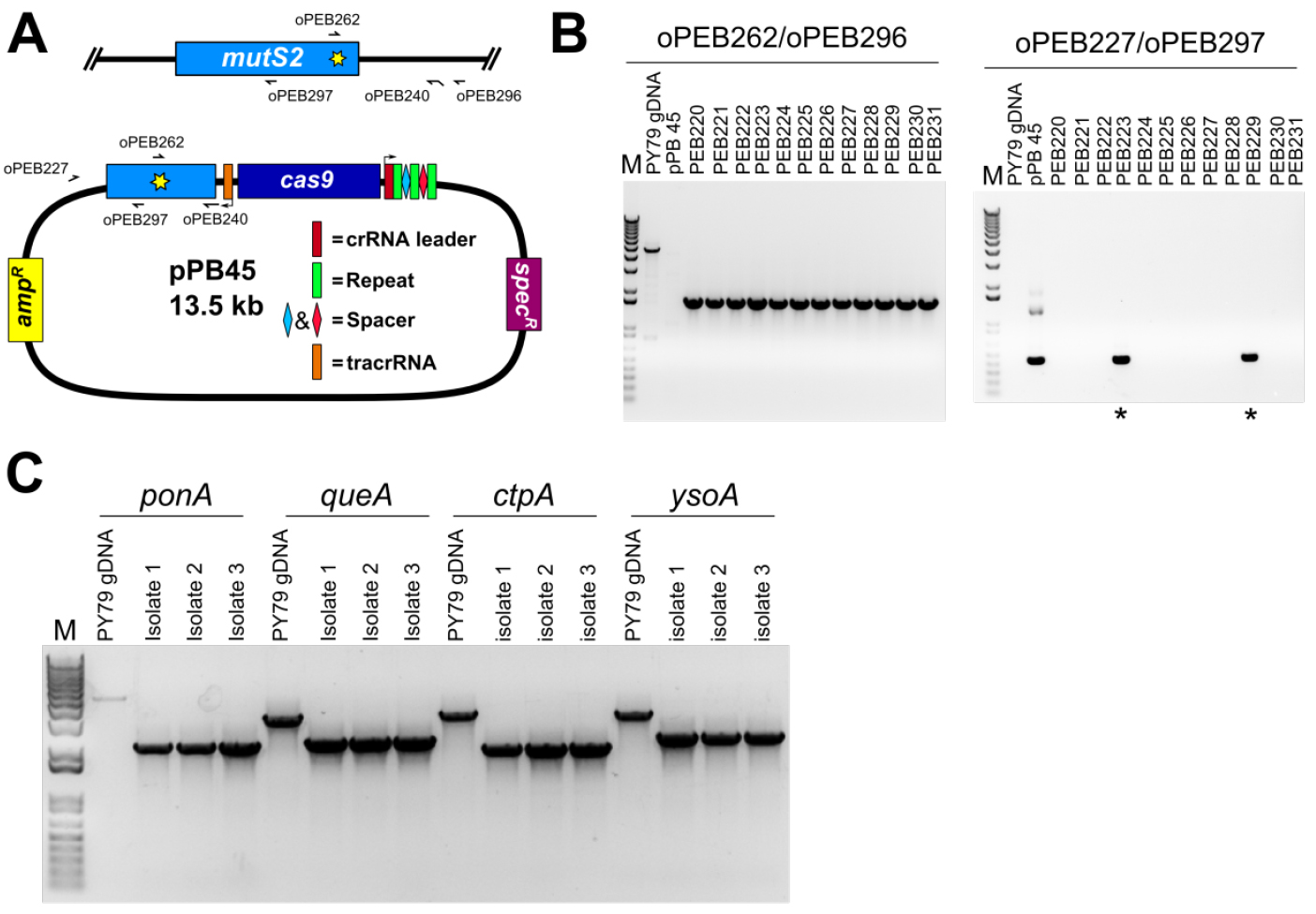
Figure 6. PCR based verification of genome edit. A. A schematic representation showing the locations of PCR primers used in panel B; B. Agarose gel stained with ethidium bromide of amplified PCR products using primers specific to the point mutation (H743A, for which we changed CAC→GCT) and the B. subtilis chromosome (left panel), and using primers specific to the editing plasmid (right panel). Of twelve isolates screened all were found to have the predicted point mutation, and two of twelve retained resistance to spectinomycin (asterisks). The slow migrating band in the PY79 lane corresponds to nonspecific PCR amplification in the wild type background. The editing plasmid was detectable in spectinomycin resistant isolates, but not in the remaining ten. C. Agarose gel stained with ethidium bromide of PCR reactions using primers specific for deletion of four B. subtilis genes. Primers were designed outside of the editing template, and are therefore specific to chromosomal DNA. For all four deletions, all isolates were found to be spectinomycin sensitive, and PCR genotyping of three isolates for each deletion found that all isolates tested had the intended deletion. Note that for the control reaction for ponA, the PCR product is faint because the amplicon is approximately 5 kb and the extension time of the PCR was 3 min, which is near the limit for complete extension for Q5 DNA polymerase; the other three control amplicons are no larger than 3.4 kb. - Save frozen stock of new strain
Separate LB cultures of at least two isolates that are confirmed to have the intended mutation are grown to an OD600 of approximately 1 to 1.5. A frozen stock is saved by adding glycerol to a final concentration of 20% in a cryo-vial. A cryo-vial cap is added to the lid and labeled appropriately. Cryo-vials are placed directly into a freezer at -80 °C.
Data analysis
There are two steps where data analysis is required for constructing a mutant strain. The first is assaying for loss of the editing plasmid using antibiotic sensitivity, and the second is to assay the genetic locus for the intended mutation using PCR genotyping. In order to determine if the isolates have been cured of the editing plasmid, six to twelve isolates are streaked onto LB agar plates with and without 100 μg/ml spectinomycin and grown overnight (see step C4 of the protocol). The isolates that do not grow on LB agar plates containing spectinomycin have been cured of the editing plasmid. The isolates that have been cured of the plasmid are further genotyped via PCR. Data analysis of PCR genotyping will depend on the type of edit being introduced. For a deletion, PCR amplification of the genetic locus will result in a product with a migration that is faster than that of the wild-type locus, resulting in the PCR product appearing lower on the agarose gel. For an insertion, the PCR product would migrate slower than the wild-type product on the agarose gel. For a point mutation, there will be no change in the size of the PCR product. As a result, a mutation specific primer or Sanger sequencing must be used to confirm the correct genotype. For a mutation specific primer (see Figure 6B), the PCR product will be present or absent. If the PCR product is present, that would indicate the mutation is present. If Sanger sequencing is used, the sequence of the mutation will be detected instead of the wild-type sequence.
Recipes
- 50x TAE
242 g Tris base
57.1 ml glacial acetic acid
100 ml EDTA, pH 8.0
Add Milli-Q H2O to 1 L
Store at room temperature - 1x TAE
20 ml 50x TAE
980 ml Milli-Q H2O
Store at room temperature - LB media
10 g NaCl
10 g tryptone
5 g yeast extract
Add Milli-Q H2O to a total volume of 1 L
Autoclave to sterilize - LB agar plates
1 L LB media
18 g agar
Autoclave to sterilize
Allow to cool to about 60 °C; if antibiotics are required, they are added at this point
Spectinomycin is added to a final concentration of 100 μg/ml
Ampicillin is added to a final concentration of 100 μg/ml
Chloramphenicol is added to a final concentration of 20 μg/ml for E. coli and 5 μg/ml for B. subtilis
Pour plates - 100 mg/ml ampicillin stock
1 g ampicillin
Add Milli-Q water to 10 ml
Filter sterilize
Aliquot and store at -20 °C - 100 mg/ml spectinomycin stock
1 g spectinomycin
Add Milli-Q water to 10 ml
Filter sterilize
Aliquot and store at -20 °C - 5 mg/ml chloramphenicol stock
50 mg chloramphenicol
Add 70% (v/v) ethanol to 10 ml
Filter sterilize
Store at -20 °C - 70% (v/v) ethanol
70 ml ethanol
30 ml Milli-Q H2O
Store at room temperature - 1% agarose gel
1 g agarose
100 mL 1x TAE
Heat solution to dissolve agarose in microwave
Add ethidium bromide to a final concentration of 0.2 μg/ml
Pour gel - QG buffer
5.5 M guanidine thiocyanate
20 mM Tris HCl, pH 6.6 - PB buffer
5 M guanidine HCl
30% (v/v) isopropanol - Tris HCl, pH 7.5
Tris HCl is prepared by dissolving Tris base in Milli-Q H2O and adjusting the pH to 7.5 using HCl - PE buffer
10 mM Tris HCl, pH 7.5
80% (v/v) ethanol - 10x annealing buffer
100 mM Tris HCl, pH 7.5
1 M NaCl
1 mM EDTA, pH 8.0
Ultra-pure H2O
Filter sterilize - 5x isothermal reaction buffer (Gibson, 2011)
0.5 M Tris HCl, pH 7.5
25% (w/v) PEG-8000
50 mM MgCl2
50 mM DTT
1 mM each of dNTPs
5 mM NAD+
Store at -20 °C - 2x Gibson master mix (Gibson, 2011)
320 μl 5x isothermal reaction buffer
0.64 μl T5 exonuclease (10,000 U/ml)
20 μl Phusion DNA polymerase (2,000 U/ml)
160 μl Taq DNA ligase (40,000 U/ml)
300 μl ddH2O
Store at -20 °C - 1 M MgSO4
120.37 g MgSO4 (F.W. 120.37 g)
Add Milli-Q H2O to 1 L
Autoclave to sterilize
Store at room temperature - LM media (LB media + 3 mM MgSO4)
2 ml LB media
6 μl 1 M MgSO4 - 10x PC buffer
10.7 g K2HPO4
6 g KH2PO4
1.18 g trisodium citrate dehydrate (C6H5Na3O7·2H2O)
Add Milli-Q H2O to 100 ml
Filter sterilize
Store at room temperature - MD media
1x PC buffer
2% (w/v) glucose
50 μg/ml tryptophan
50 μg/ml phenylalanine
11 μg/ml ferric ammonium citrate
2.5 mg/ml potassium aspartate
3 mM MgSO4
Filter sterilize
Store protected from light at 4 °C - 0.1 N KOH
56.1 mg KOH (F.W. 56.11 g)
Add Milli-Q H2O to 10 ml
Prepare fresh for each use - 100 mg/ml tryptophan stock
1 g tryptophan
Add 0.1 N KOH to 10 ml (prepare fresh)
Filter sterilize
Store protected from light at 4 °C - 100 mg/ml phenylalanine stock
1 g phenylalanine
Add 0.1 N KOH to 10 ml (prepare fresh)
Filter sterilize
Store protected from light at 4 °C - 2.2 mg/ml ferric ammonium citrate stock
110 mg ferric ammonium citrate
Add Milli-Q H2O to 50 ml
Filter sterilize
Store protected from light at 4 °C - 100 mg/ml potassium aspartate stock
5 g potassium aspartate
Add Milli-Q H2O to 50 ml
Filter sterilize
Store at 4 °C
Acknowledgments
This work was supported by NIH grant R01 GM107312 to L.A.S. and a pre-doctoral fellowship from the National Science Foundation #DGE 1256260 to P.E.B.
References
- Arnaud, M., Chastanet, A. and Debarbouille, M. (2004). New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, Gram-positive bacteria. Appl Environ Microbiol 70(11): 6887-6891.
- Burby, P. E. and Simmons, L. A. (2017). MutS2 promotes homologous recombination in Bacillus subtilis. J Bacteriol 199(2).
- Fabret, C., Ehrlich, S. D. and Noirot, P. (2002). A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol 46(1): 25-36.
- Gibson, D. G. (2011). Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498: 349-361.
- Guerout-Fleury, A. M., Frandsen, N. and Stragier, P. (1996). Plasmids for ectopic integration in Bacillus subtilis. Gene 180(1-2): 57-61.
- Jiang, W., Bikard, D., Cox, D., Zhang, F. and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31(3): 233-239.
- Kibbe, W. A. (2007). OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res 35(Web Server issue): W43-46.
- Koressaar, T. and Remm, M. (2007). Enhancements and modifications of primer design program Primer3. Bioinformatics 23(10): 1289-1291.
- Patrick, J. E. and Kearns, D. B. (2008). MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol Microbiol 70(5): 1166-1179.
- Sternberg, S. H. and Doudna, J. A. (2015). Expanding the biologist's toolkit with CRISPR-Cas9. Mol Cell 58(4): 568-574.
- Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M. and Rozen, S. G. (2012). Primer3--new capabilities and interfaces. Nucleic Acids Res 40(15): e115.
- Vagner, V., Dervyn, E. and Ehrlich, S. D. (1998). A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144: 3097-3104.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Burby, P. E. and Simmons, L. A. (2017). CRISPR/Cas9 Editing of the Bacillus subtilis Genome. Bio-protocol 7(8): e2272. DOI: 10.21769/BioProtoc.2272.
Category
Microbiology > Microbial genetics > DNA > Chromosomal
Microbiology > in vivo model > Bacterium
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.