- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Use of Geminivirus for Delivery of CRISPR/Cas9 Components to Tobacco by Agro-infiltration


Published: Vol 7, Iss 7, Apr 5, 2017 DOI: 10.21769/BioProtoc.2209 Views: 15888
Reviewed by: Rainer MelzerKaisa KajalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
CRISPR/Cas9 system is a recently developed genome editing tool, and its power has been demonstrated in many organisms, including some plant species (Wang et al., 2016). In eukaryotes, the Cas9/gRNA complexes target genome sites specifically and cleave them to produce double-strand breaks (DSBs), which can be repaired by non-homologous end joining (NHEJ) pathway (Wang et al., 2016). Since NHEJ is error prone, mutations are thus generated. In plants, delivery of genome editing reagents is still challenging. In this protocol, we detail the procedure of a virus-based gRNA delivery system for CRISPR/Cas9 mediated plant genome editing (VIGE). This method offers a rapid and efficient way to deliver gRNA into plant cells, especially for those that are recalcitrant to transformation with Agrobacterium.
Keywords: CRISPRBackground
Genome editing technologies based on viruses have been reported using deconstructed DNA viruses and an RNA virus (Baltes et al., 2014; Ali et al., 2015). Recently, we used a full geminivirus - Cabbage Leaf Curl virus (CaLCuV) (a bipartite begomovirus which infects a wide range of members of the Brassicaceae, including cauliflower) for highly efficient genome editing in one of its hosts, Nicotiana benthamiana, for the first time (Yin et al., 2015).
Materials and Reagents
- 1-ml syringe
- 0.22-μm filter
- Wild type N. benthamiana plants; six to eight-leaf stage transgenic N. benthamiana plants stably expressing oCas9 (Arabidopsis codon optimized Cas9) - KQ334 plant
- Agrobacterium tumefaciens strain GV3101, E. coli DH5α, E. coli DB3.1 (for propagating vectors pJG081)
- The following plasmids are required: pJG081, pKQ334 (Yin et al., 2015), pT-U6p-scaffold-U6t (Yin et al., 2015), pCAMBIA1301, pCAMBIA2300 (www.cambia.org), pCVA (Tang et al., 2010), pCVB (Tang et al., 2010), and pMD18-T vector (Takara Bio, catalog number: D101A)
Notes: - To generate a construct for constitutive expression of oCas9, oCas9 is PCR amplified and subsequently cloned into pJG081 by ligation independent cloning (LIC) technique (Aslanidis and de Jong, 1990) to generate pKQ334.
- pKQ334 is then used to transform N. benthamiana to generate Cas9 transgenic plant - KQ334 plant. pKQ334 can also be used in transient expression of Cas9 in N. benthamiana.
- The pT-U6p-scaffold-U6t is used to generate p-T-U6p-gRNA-scaffold containing gRNA of the target gene by PCR mutagenesis.
- pCVA and pCVB are CaLCuV-based T-DNA vectors. pCVA is used to generate pCVA-gRNA or pCVA-scaffold. pCVB is used in combination with pCVA or its derivatives to generate CaLCuV. pMD18-T vector is used to clone PCR amplicons.
- These vectors can be obtained by contacting Dr. Yule Liu.
- Primers (Table 1)
Table 1. Primers used in this protocol
- TransStart FastPfu DNA polymerase (Beijing TransGen Biotech, catalog number: AP221 )
- Restriction enzymes (New England Biolabs, stored at -20 °C)
- DpnI (New England Biolabs, catalog number: R0176 )
- ApaI (New England Biolabs, catalog number: R0114 )
- SacI (New England Biolabs, catalog number: R0156 )
- PstI (New England Biolabs, catalog number: R3140S )
- KpnI (New England Biolabs, catalog number: R3142S )
- XbaI (New England Biolabs, catalog number: R0145S )
- MlyI (New England Biolabs, catalog number: R0610 )
- 10x Cutsmart buffer
- Antibiotics
Ampicillin (AMRESCO, catalog number: 0339 )
Kanamycin (AMRESCO, catalog number: 0408 )
Rifampicin (Sigma-Aldrich, catalog number: R3501 ) - TIANprep Mini Plasmid Kit (TIANGEN Biotech, catalog number: DP103 ) for plasmid DNA extraction
- Ethanol (Beijing Chemical Factory, catalog number: B0301002 )
- T4 DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0062 )
- dATP (Takara Bio, catalog number: 4026 )
- Chloroform (Sigma-Aldrich, catalog number: 1601383 )
- Phenol (Sigma-Aldrich, catalog number: P4557 )
- 3 M NaAc (Biovision, catalog number: 2118 )
- dTTP (Takara Bio, catalog number: 4029 )
- DNA Purification Kit (BioMED, catalog number: DH103-01 )
- T4 DNA ligase (Takara Bio, catalog number: 2011A )
- Tryptone (BD, BactoTM, catalog number: 211705 )
- Yeast extract (BD, BactoTM, catalog number: 212750 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 31434 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M7304 )
- MES (Sigma-Aldrich, catalog number: M3671 )
- Potassium hydroxide (KOH)
- Acetosyringone (Solarbio, catalog number: A8110-1 )
- DMSO
- Sodium phosphate monobasic (NaH2PO4) (Sigma-Aldrich, catalog number: S3139 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S3264 )
- Sodium phosphate monobasic (NaH2PO4) (AMRESCO, catalog number: 0571 )
- Sodium phosphate dibasic (Na2HPO4) (AMRESCO, catalog number: 0404 )
- Sodium phosphate monobasic monohydrate (NaH2PO4·H2O)
- Sodium phosphate dibasic heptahydrate (Na2HPO4·7H2O)
- X-gluc (AMRESCO, catalog number: 0919 )
- Methanol (Beijing Chemical Factory, catalog number: B0301005 )
- Potassium ferricyanide (AMRESCO, catalog number: 0713 )
- Triton X-100 (Sigma-Aldrich, catalog number: T9284 )
- PEG 8000 (Sigma-Aldrich, catalog number: 89510 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (AMRESCO, catalog number: 0288 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: 63068 )
- MES (Sigma-Aldrich, catalog number: M3671 )
- Acetosyringone (Sigma-Aldrich, catalog number: D134406 )
- EasyTaq DNA polymerase (Beijing TransGen Biotech, catalog number: AP111 )
- DNAsecure Plant Kit (TIANGEN Biotech, catalog number: DP320 ) for genomic DNA extraction
- Luria-Bertani medium (see Recipes)
- MgCl2 (1 M stock) (see Recipes)
- MES (0.5 M stock) (see Recipes)
- Acetosyringone (1 M stock) (see Recipes)
- Infiltration buffer (see Recipes)
- 2x phosphate buffer (pH 7) (see Recipes)
- X-gluc substrate solution (see Recipes)
- PEG-MgCl2 solution (see Recipes)
Equipment
- Microcentrifuge (Eppendorf)
- PCR cycler (Bio-Rad Laboratories)
- 37 °C and 28 °C incubator; 37 °C and 28 °C shaker
- NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, model: NanoDrop 1000 ) (for measuring the concentration of the DNA)
- Electrophoresis apparatus (Junyi, Beijing)
- Vacuum apparatus (WILMAD-LABGLASS)
- Glass/plastic beaker
- Autoclave
Software
- ImageJ (https://imagej.nih.gov/ij)
Procedure
- gRNA design for specific gene/site of interest
For knocking out a gene of interest, choose gRNAs located in the exons. For editing the non CDS region in the genome, choose a region where disruption would have this desired effect. We strongly recommend using CRISPR-P (cbi.hzau.edu.cn) to design gRNAs (Lei et al., 2014). It provides a specificity score for each possible gRNA and lists the potential off-targets. Additionally, it provides a list of useful restriction enzymes that would recognize the gRNA spacer for RFLP analysis of this specific site. - Construct gRNA plasmid
To drive gRNA expression in plants, we constructed a gRNA plasmid targeting NbPDS. Any other genes-of-interest can also be targeted. To clone the gRNA easily, we first used a small vector (T-U6p-scaffold-U6t). Then, the promoter-gRNA-terminator construct is transferred to a larger binary vector, such as pCAMBIA2300 and pCVA.- Perform inverse PCR (Stemmer and Morris, 1992) to add gRNA spacer between the AtU6 promoter and gRNA scaffold necessary for Cas9 binding (Figure 1). Setup reactions as follows:

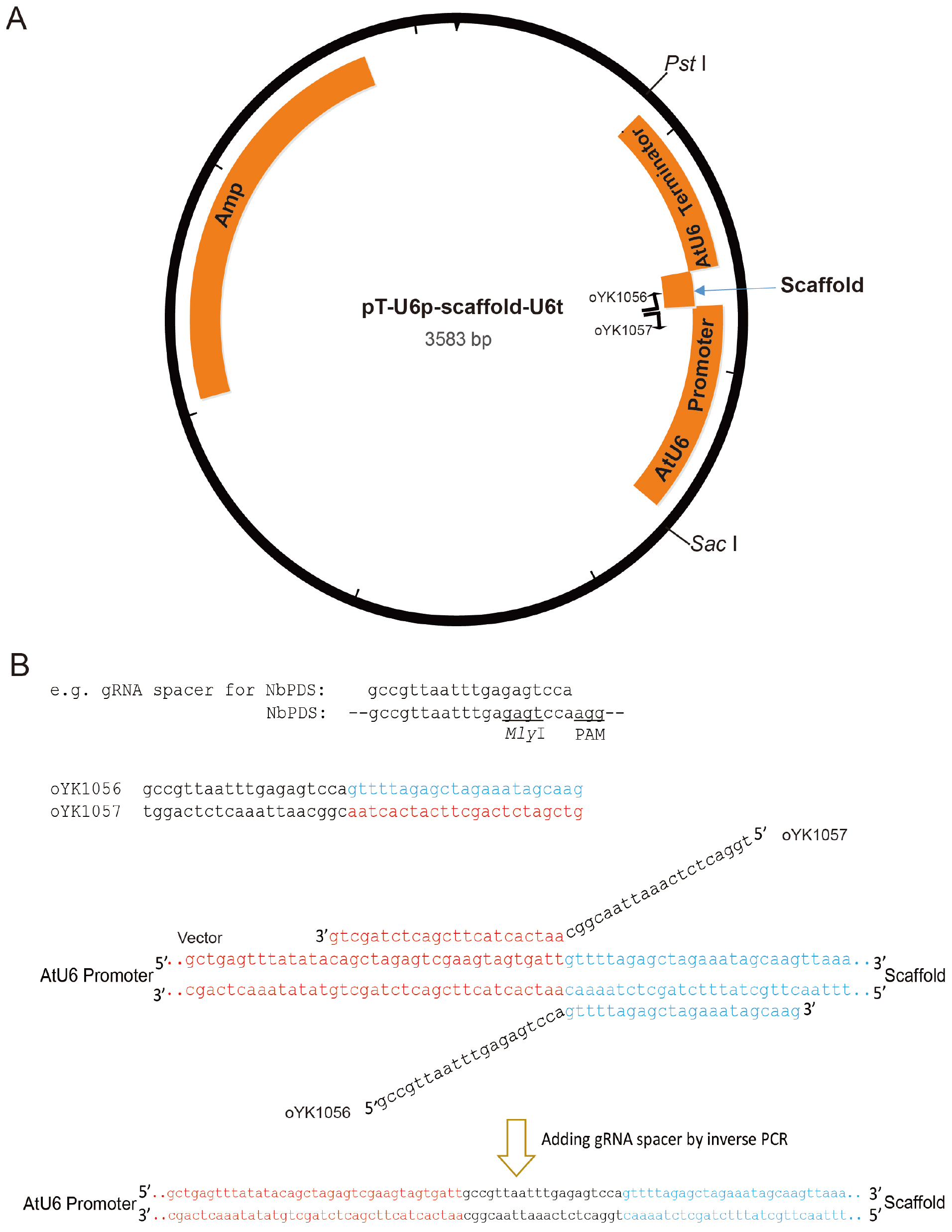
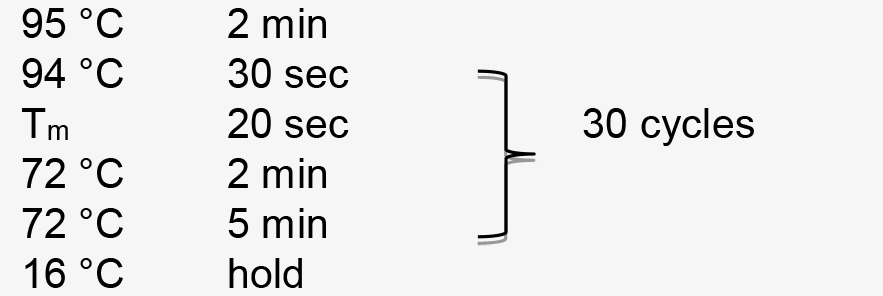
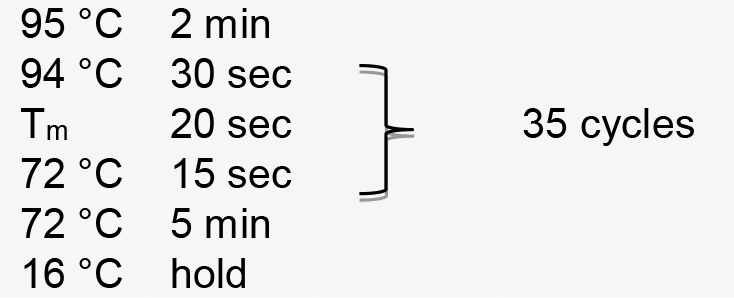
Figure 1. Illustration of pT-U6p-scaffold-U6t (Yin et al., 2015) and the use of inverse PCR for inserting gRNA spacer into pT-U6p-scaffold-U6t to generate pT-U6p-gRNA-scaffold-U6t. A. AtU6 promoter, scaffold, and AtU6 terminator are shown in orange boxes on the plasmid map. Two primers used for inserting gRNA spacer (targeting NbPDS) are indicated on the plasmid map with their respective directions. Parallel blacklines on the primers are complementary sequences. SacI and PstI are used to cut the gRNA expression cassette which is then cloned into a binary vector. The small arrow here is to indicate scaffold. B. Sequence information for inverse PCR. Here, gRNA spacer targeting NbPDS is used as an example. Partial sequence of the NbPDS locus is shown in black. PAM and MlyI site are shown. gRNA spacer is also shown in black above the NbPDS locus. Two primers (oYK1056 and oYK1057) for PCR are shown, the sequence from AtU6 is shown in red, and the sequence from scaffold is shown in blue. The two primers that bind to the template (vector) during the PCR process are also shown with primers and vector indicated. The change after inverse PCR is shown below the arrow. - Perform PCR amplification using the following parameters (Tm represents the annealing temperature of the primer pair):
- Setup a restriction enzyme digest to remove the plasmid template in the above PCR product as follows:
Note: DpnI is a methylation sensitive enzyme, and plasmid purified from E. coli DH5α is generally methylated. Thus, DpnI is often used to remove plasmid template. Here, a ligation or a denaturation/reannealing step is not necessary to connect the ends of the vector, because the PCR product with overlapping ends will re-circularize to form a doubly-nicked plasmid and will be repaired further in bacteria to form an intact plasmid.
Mix well and incubate at 37 °C for 2 h. - Transform E.coli DH5α using 1-2 μl product of the above reaction with heat shock at 42 °C for 90 sec, recover at 37 °C for 1 h, plate on LB plate with 100 mg/L ampicillin, and then incubate overnight at 37 °C (Im, 2011).
- Select one to five transformants and culture overnight in LB with 100 mg/L ampicillin at 37 °C, 220 rpm. Extract plasmids from the transformants respectively with TIANprep Mini Plasmid Kit, and Sanger sequence the plasmid pT-U6p-gRNA:NbPDS-scaffold-U6t with sequencing primer M13-47 to confirm the presence of the gRNA spacer.
- Perform inverse PCR (Stemmer and Morris, 1992) to add gRNA spacer between the AtU6 promoter and gRNA scaffold necessary for Cas9 binding (Figure 1). Setup reactions as follows:
- Check Cas9 activity and gRNA efficiency using fsGUS (frameshift GUS) reporter system
It’s necessary to confirm if KQ334 plants retain Cas9 activity and that Cas9 cleaves DNA under the guidance of gRNA. In this system, a 23 bp sequence with a PAM (AGG) at the end is inserted downstream of the start codon of the GUS gene, which causes a frame shift of the GUS gene (fsGUS). We co-expressed two vectors: one harboring the fsGUS gene driven by a 35S promoter termed pJG081-fsGUS and the other p2300-gRNA:fsGUS containing an Arabidopsis U6-26 gene promoter (AtU6 promoter)-driven gRNA containing a 20 bp sequence that matches the first 20 bp of the sequence inserted into the fsGUS gene.
There is another highly efficient method to check gRNA efficiency based on protoplast transfection. Please refer to Li and co-workers’ protocol (Li et al., 2015)- Setup a PCR reaction to amplify an fsGUS fragment in which a short sequence containing the gRNA spacer sequence and a MlyI recognition site is inserted after the GUS start codon (Figure 2). Primers used at this step should be designed specifically to flank the cut sites of different gRNAs.

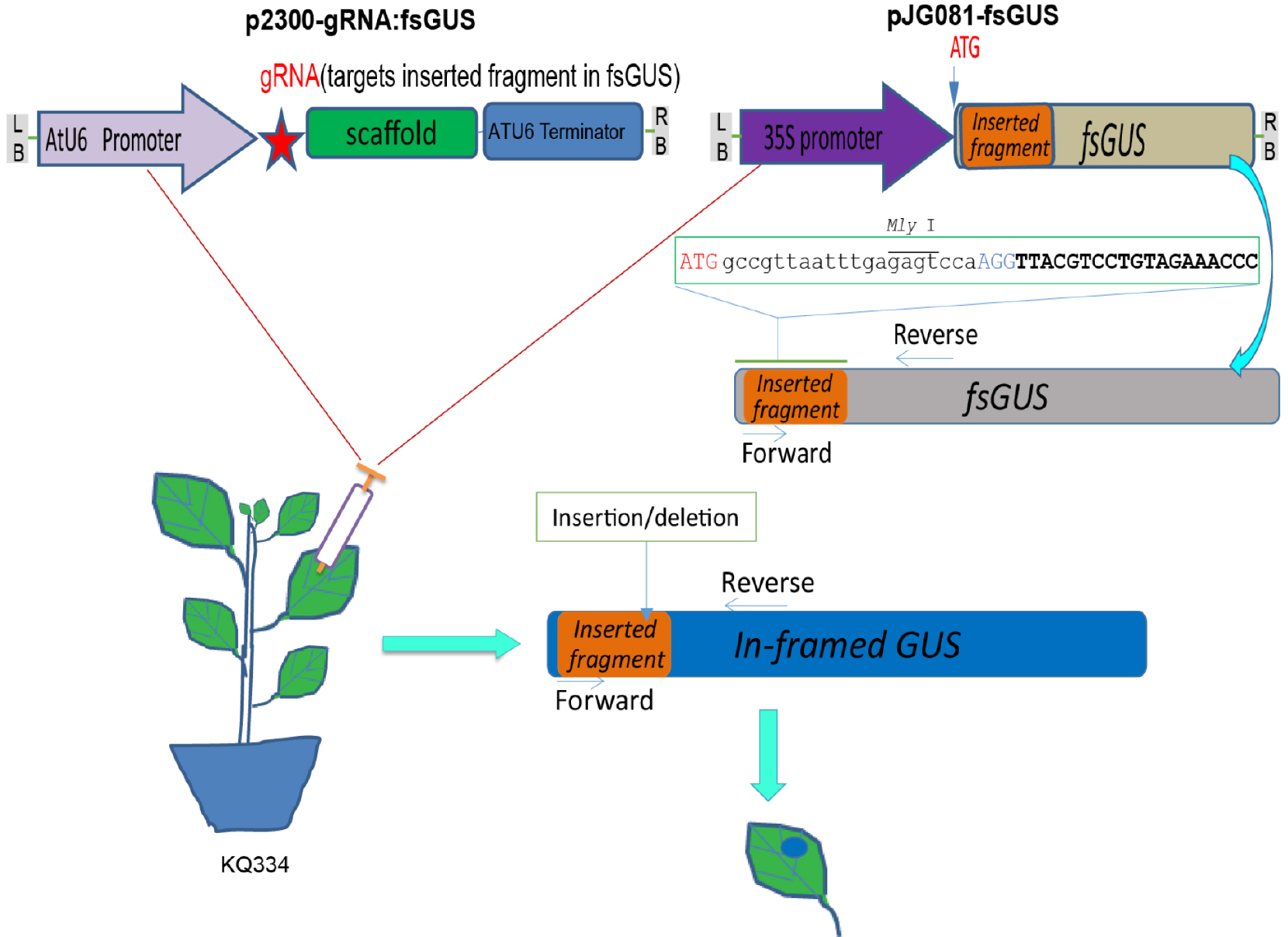 Figure 2. Illustration of the fsGUS reporter system used to check gRNA efficiency. Illustration of the fsGUS reporter system. A 23 bp sequence containing a MlyI site is inserted after the start codon of the GUS gene, and the resulting sequence is designated as fsGUS. Due to the inserted sequence, fsGUS cannot be translated into a functional enzyme. fsGUS is driven by a CaMV35S promoter and placed in a binary vector. Another binary vector contains AtU6 promoter-driven gRNA that targets the first 20 bp of the inserted sequence in fsGUS. Two A. tumefaciens lines transformed with each binary vector are mixed together and infiltrated into KQ334 plant leaves. After two T-DNAs enter into the same cell, the first 20 bp of the inserted sequence in fsGUS is targeted by the Cas9/gRNA complex for cleavage. Repair via NHEJ will create in-dels at the cleavage site and, in some cases, recover the normal coding frame for fsGUS, thus creating a functional GUS which can be detected by GUS staining. (This figure is adapted and modified from Yin et al., 2015.)Perform PCR amplification using the following parameters (Tm represents the annealing temperature of the primer pair):
Figure 2. Illustration of the fsGUS reporter system used to check gRNA efficiency. Illustration of the fsGUS reporter system. A 23 bp sequence containing a MlyI site is inserted after the start codon of the GUS gene, and the resulting sequence is designated as fsGUS. Due to the inserted sequence, fsGUS cannot be translated into a functional enzyme. fsGUS is driven by a CaMV35S promoter and placed in a binary vector. Another binary vector contains AtU6 promoter-driven gRNA that targets the first 20 bp of the inserted sequence in fsGUS. Two A. tumefaciens lines transformed with each binary vector are mixed together and infiltrated into KQ334 plant leaves. After two T-DNAs enter into the same cell, the first 20 bp of the inserted sequence in fsGUS is targeted by the Cas9/gRNA complex for cleavage. Repair via NHEJ will create in-dels at the cleavage site and, in some cases, recover the normal coding frame for fsGUS, thus creating a functional GUS which can be detected by GUS staining. (This figure is adapted and modified from Yin et al., 2015.)Perform PCR amplification using the following parameters (Tm represents the annealing temperature of the primer pair):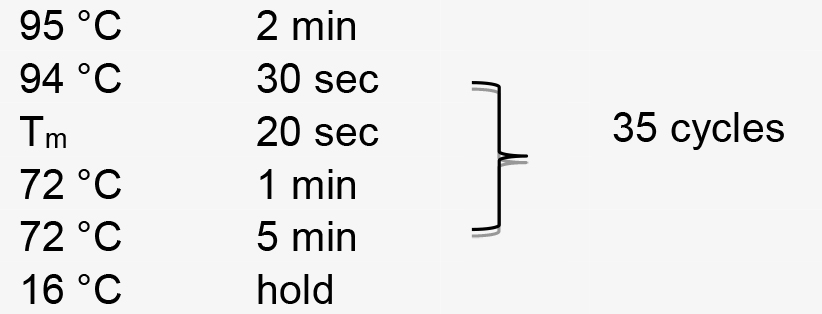
- Purify the PCR product using PEG/MgCl2 method (Paithankar and Prasad, 1991)
- Add 50 μl water to 50 μl PCR product and then add 50 μl 30% PEG8000/30 mM MgCl2.
- Mix well and centrifuge immediately at 12,000 x g for 15 min.
- Next, remove the supernatant and add 200 μl 70% ethanol.
- Centrifuge at 12,000 x g for 5 min and remove the supernatant. Finally, dry the pellet and dissolve the pellet in 10 μl sterile deionized water.
- Construct pJG081-fsGUS using LIC method.
Note: LIC is a fast, simple, and very cheap cloning technique. It utilizes annealing of complementary overhangs of a vector and a PCR fragment consisting of 9-15 bases. - Single-stranded overhangs are generated by using T4 DNA polymerase and the addition of only one dNTP in the reaction buffer, leading to an equilibrium of activity of polymerase and exonuclease at the first appearance of this nucleotide (Aslanidis and de Jong, 1990). For the generation of the single stranded tails, the purified DNA preparations are treated with T4 DNA polymerase. Setup T4 DNA polymerase treatment for the above PCR product:

- Mix gently and well by pipetting. Incubate at 37 °C for 20 min then at 75 °C for 20 min to inactivate T4 DNA polymerase using a PCR cycler. The resulting product is designated as fsGUS-LIC.
- Digest pJG081 for LIC reaction:

- Incubate at 37 °C in incubator for 3 h.
- Then, purify the product using phenol/chloroform method: add an equal volume of phenol/chloroform (1:1) and vortex vigorously for 20 sec; then centrifuge at 12,000 x g for 10 min at room temperature; retain the supernatant and add an equal volume of chloroform, vortex vigorously for 20 sec, and centrifuge at 12,000 x g for 10 min at room temperature; precipitate the DNA from the aqueous phase of the above extractions by adding 1/10 volume of 3 M NaAc and 2 volume of ethanol.
- Finally, pellet the DNA by centrifugation, and dissolve it with sterile deionized water.
- Setup T4 DNA polymerase treatment for this linear pJG081:

The resulting product is designated as pJG081-LIC. - Construct pJG081-fsGUS:
- Mix 5 μl fsGUS-LIC and 5 μl pJG081-LIC well, incubate at 70 °C for 10 min and then at 22 °C for 10 min.
- Next, transform this product into E. coli DH5α, which is then selected on medium with kanamycin.
- Construct p2300-gRNA:fsGUS.
- Setup a restriction enzyme reaction as follows:

- Incubate at 37 °C for 3 h. Then, purify the product with the DNA Purification Kit according to manufacturer recommendations.
- To clone a complete gRNA expression cassette into pCambia 2300, U6p-gRNA:NbPDS-scaffold-U6t is cut from pT-U6p-gRNA:NbPDS-scaffold-U6t. Setup a restriction enzyme reaction as follows:

- Incubate at 37 °C for 3 h. Then purify the product with the DNA Purification Kit according to manufacturer recommendations.
- Setup a ligation reaction as follows:
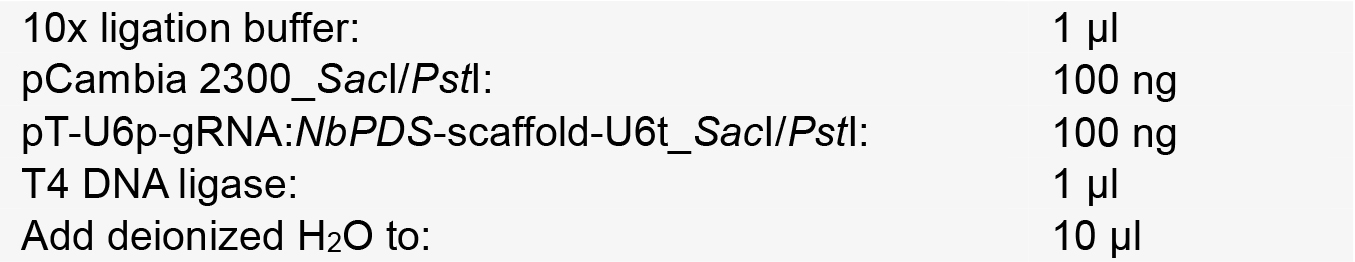
- Incubate at 16 °C for 8 h and transform it into E. coli DH5α, which is then selected on medium with kanamycin.
- Agrobacterium transformation
- Transform pKQ334, pJG081-fsGUS and p2300-gRNA:fsGUS respectively into Agrobacterium tumefaciens strain GV3101 by heat shock method (Höfgen and Willmitzer, 1988).
- Select the transformants on LB medium with 50 mg L-1 kanamycin and 50 mg L-1 rifampicin at 28 °C for two days.
- Transient expression
Note: Either Cas9 transgenic plants (KQ334 plants) or another Agrobacterium culture containing pKQ334 plasmid (constitutive expression of Cas9) can be used for this experiment. - The Agrobacterium cultures are inoculated in a 5 ml Luria-Bertani medium containing appropriate antibiotics (50 mg L-1 rifampicin, and 50 mg L-1 kanamycin) and grown overnight in a 28 °C shaker.
- Agrobacterium cells are harvested by centrifugation at 5,000 x g for 5 min and resuspended in infiltration buffer, adjusted to an optical density at 600 nm of 1.0 and incubated at room temperature for 3 to 4 h before infiltration.
- For transient expression, equal amounts of Agrobacterium strain GV3101 cultures containing either pKQ334 or pJG081-fsGUS or p2300-gRNA:fsGUS are mixed as experimental group and spot-infiltrated into leaves of six-leaf stage plants using a 1-ml syringe. As control, Agrobacterium cultures containing either pJG081-fsGUS or p2300-gRNA:fsGUS are spot-infiltrated separately into leaves of six-leaf stage plants using a 1-ml syringe.
- The first three leaves counting from the top of each plant are infiltrated; each leaf is spot-infiltrated with experimental group and control group. Midrib and main veins are avoided when infiltrating.
- Press the nozzle of a 1 ml syringe without needle against the abaxial surface of the leaf, and hold a gloved finger on the other side and inject slowly. The infiltration area is often seen as ‘wet’ (Video 1).
 Video 1. Transient expression using agro-infiltration in N. benthamiana
Video 1. Transient expression using agro-infiltration in N. benthamiana - Alternatively, equal amounts of Agrobacterium cultures containing either pJG081-fsGUS or p2300-gRNA:fsGUS can be mixed together and infiltrated into leaves of six-leaf stage KQ334 plants using a 1-ml syringe (Figure 2). As control, Agrobacterium cultures containing either pJG081-fsGUS or p2300-gRNA:fsGUS are infiltrated separately into leaves of six-leaf stage KQ334 plants using a 1-ml syringe.
- GUS staining
The GUS staining assay is based on Kabbage et al. (2011) and performed as follows: - Two to three days after infiltration, infiltrated leaves are collected. Vacuum infiltrate the X-gluc substrate into the collected leaves. A glass/plastic beaker is used to put leaves in for vacuum infiltration and a vacuum level of 600 mm Hg is applied (for 3 min) and released for several times until the leaves become translucent.
- After infiltration, leaves are incubated in darkness at room temperature overnight.
- Rinse the leaves by changing the solution with deionized water and remove the chlorophyll by immersing the leaves in 70% ethanol for at least one day.
- Setup a PCR reaction to amplify an fsGUS fragment in which a short sequence containing the gRNA spacer sequence and a MlyI recognition site is inserted after the GUS start codon (Figure 2). Primers used at this step should be designed specifically to flank the cut sites of different gRNAs.
- Construct pCVA-gRNA and pCVA-scaffold
To perform VIGE of the target gene, pCVA-gRNA is first generated by inserting a cassette containing an AtU6 promoter and gRNA::NbPDS into pCVA. The construct pCVA-scaffold is taken as a negative control.- Setup PCR reactions as follows:

- Perform PCR amplification using the following parameters (Tm represents the annealing temperature of the primer pair):
- Then purify the product with the DNA Purification Kit according to manufacturer recommendations.
- Setup a restriction enzyme reaction as follows:

- Incubate at 37 °C for 3 h. Then purify the product with the DNA Purification Kit. The final products are U6p-gRNA-scaffold_KpnI/XbaI and U6p-scaffold_KpnI/XbaI.
- At the same time, setup a restriction enzyme reaction as follows:

- Incubate at 37 °C for 3 h. Then purify the product with the DNA Purification Kit according to manufacturer recommendations.
- Next, setup a ligation reaction as follows to generate pCVA-gRNA and pCVA-scaffold

- Incubate at 16 °C for 8 h and transform it into E. coli DH5α, which is then selected on medium with kanamycin.
- Setup PCR reactions as follows:
- VIGE of a gene/site of interest
- First, transform pCVA-gRNA, pCVA-scaffold and pCVB respectively into Agrobacterium tumefaciens strain GV3101 by heat shock method. Select the transformants on medium with kanamycin and rifampicin.
- Next, the Agrobacterium cultures are inoculated to 5 ml Luria-Bertani medium containing appropriate antibiotics (50 mg L-1 rifampicin, and 50 mg L-1 kanamycin) and grown overnight in a 28 °C shaker.
- Agrobacterium cells are harvested by centrifugation at 5,000 x g for 5 min and resuspended in infiltration buffer, adjusted to an optical density at 600 nm of 1.0 and incubated at room temperature for 3 to 4 h before infiltration.
- Agrobacterium cultures containing pCVB and pCVA or their derivatives are mixed at a 1:1 ratio and infiltrated into petioles of six to eight-leaf stage KQ334 plants using a 1-ml syringe (Tang et al., 2010) (Figure 3 and Video 2). As a control, they are also infiltrated into petioles of six to eight-leaf stage wild-type plants using the same method.
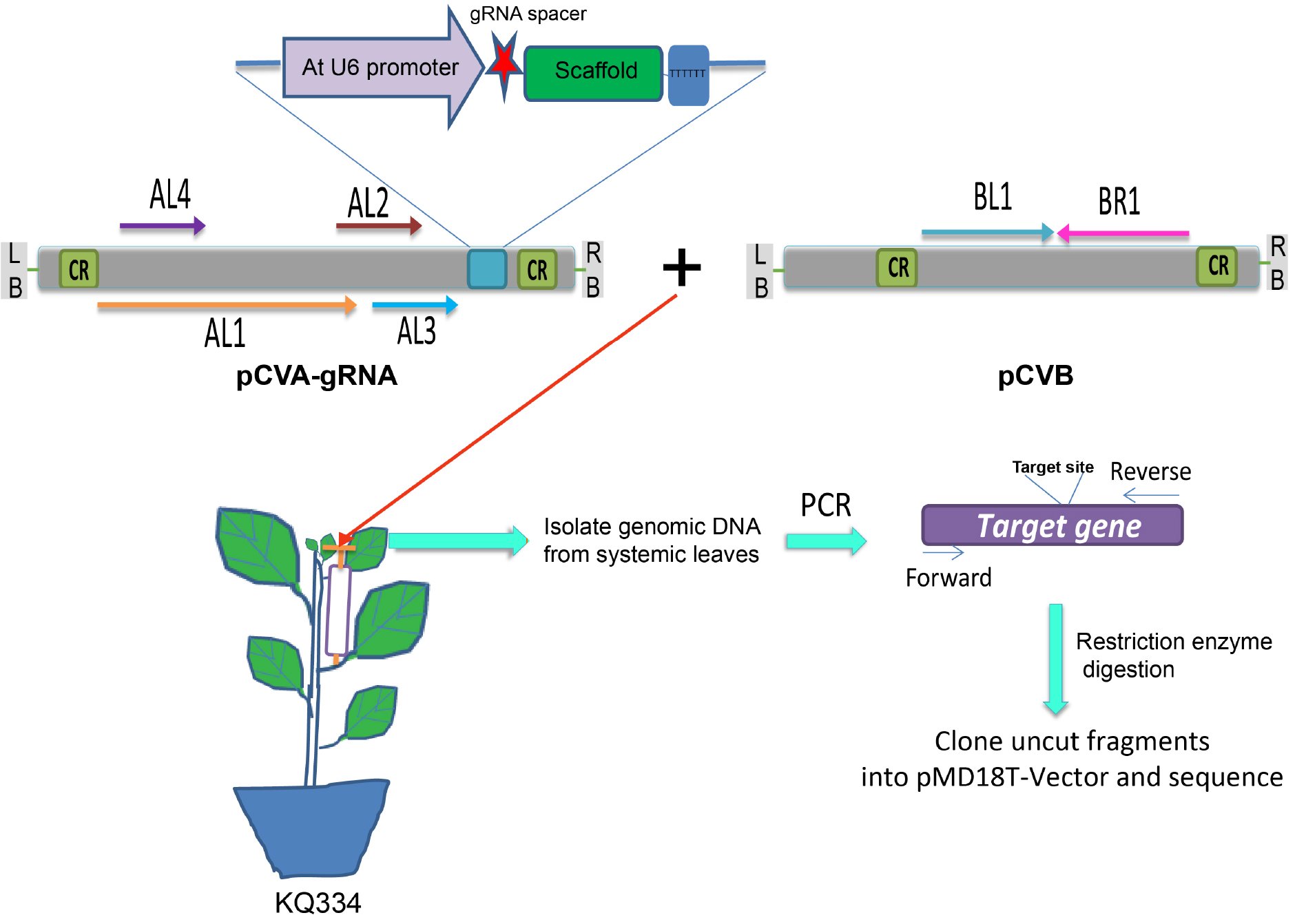
Figure 3. Illustration of VIGE. The Agrobacterium containing either pCVA-gRNA or pCVB are mixed with each other and infiltrated into petiole of KQ334 plants (Cas9 transgenic plants). Systemic leaves are used to check the genome editing events. AtU6 promoter is used to drive the expression of gRNA. The gRNA spacer is shown in red. CR, common region; AL1, replication-associated protein; AL2, transcription activator; AL3, replication enhancer; AL4, putative pathogenesis-related protein; BL1 and BR1, movement proteins; LB, left border; RB, right border. (This figure is adapted and modified from Yin et al., 2015.) Video 2. Agro-infiltration into petiole of N. benthamiana
- First, transform pCVA-gRNA, pCVA-scaffold and pCVB respectively into Agrobacterium tumefaciens strain GV3101 by heat shock method. Select the transformants on medium with kanamycin and rifampicin.
- Detection of VIGE mediated mutations in plant genomic DNA
- For the detection of VIGE results, genomic DNA extracted from systemic leaves according to the manufacturer’s instructions is used as template for PCR amplification (see below) of a targeted locus with primers (PDS_MlyIF and PDS_MlyIR) flanking the target site using EasyTaq DNA polymerase. PCR product is purified and digested with selected restriction enzyme (i.e., MlyI for NbPDS) and run on a 2% agarose gel.
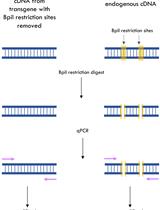
- Mutation rate is estimated by comparing band intensities using ImageJ (https://imagej.nih.gov/ij). The uncut band is then cut from the gel and the DNA is recovered by using DNA Purification Kit according to the manufacturer’s instructions. The undigested purified DNA is subsequently cloned into pMD18-T vector (TAKARA) and transformed into E. coli DH5α. Clones are sequenced with M13-47 primer to check for mutations (Figure 3). An example of VIGE for NbPDS gene is shown in Figure 4, in which the PCR product of a specific primer pair targeting the NbPDS gene is digested with MlyI.
PCR amplification is performed using the following parameters (Tm represents the annealing temperature of the primer pair):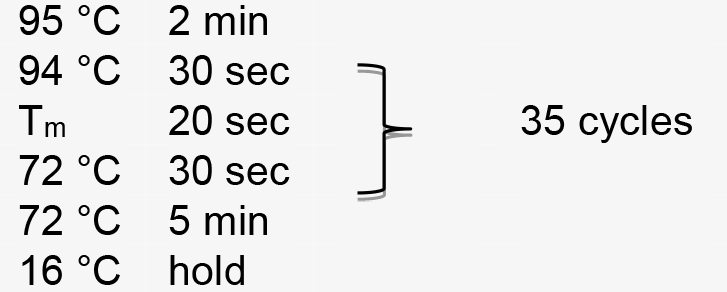
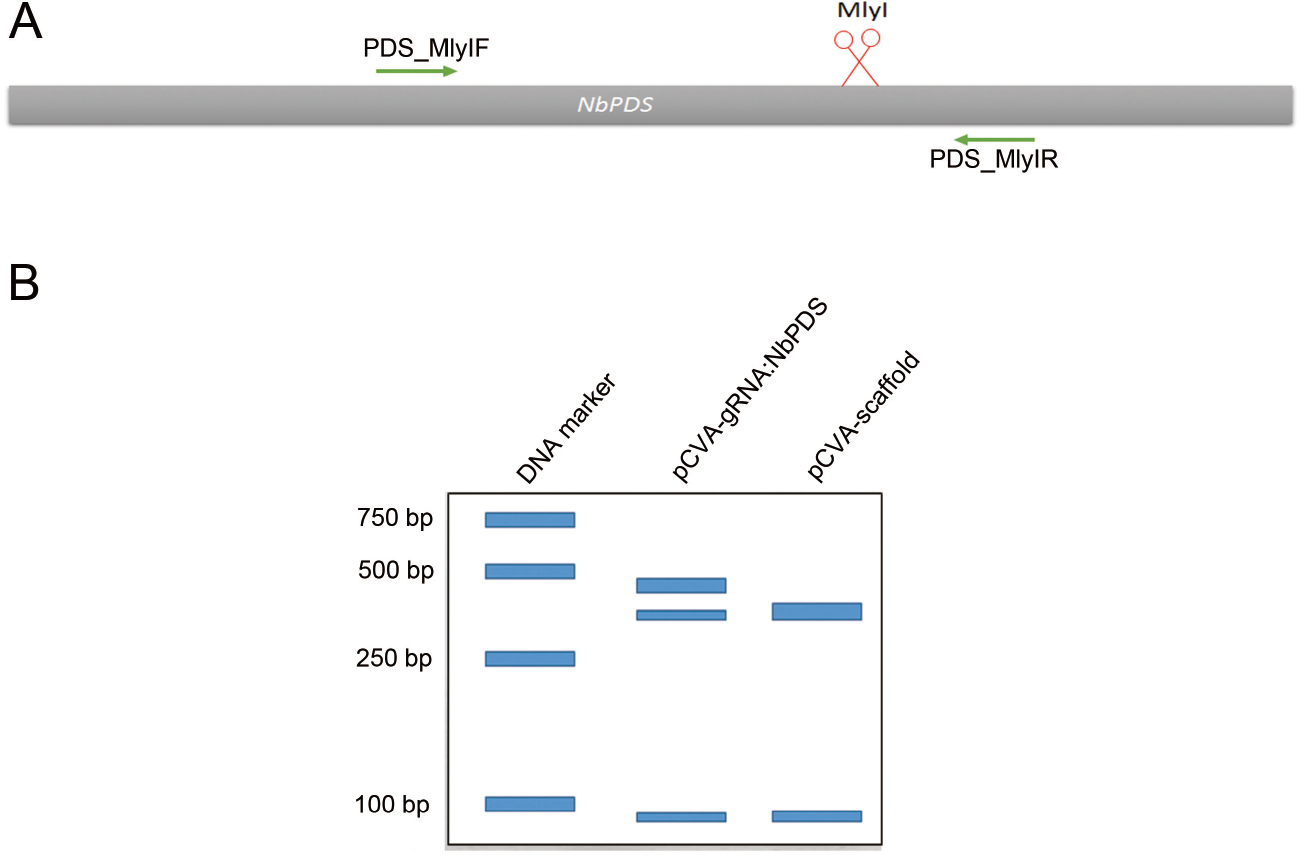
Figure 4. VIGE of NbPDS. A. illustration of VIGE targeted gene NbPDS. Cut site of Cas9/gRNA is under the red scissor. MlyI is used for discriminating the edited and wild-type sequence. B. schematic of the gel assay used to estimate the editing efficiency of VIGE of NbPDS gene.
- For the detection of VIGE results, genomic DNA extracted from systemic leaves according to the manufacturer’s instructions is used as template for PCR amplification (see below) of a targeted locus with primers (PDS_MlyIF and PDS_MlyIR) flanking the target site using EasyTaq DNA polymerase. PCR product is purified and digested with selected restriction enzyme (i.e., MlyI for NbPDS) and run on a 2% agarose gel.
Data analysis
To investigate whether endogenous NbPDS gene is edited, ImageJ is used to evaluate the editing efficiency by VIGE.
- Select Lanes for analysis and add frames to include bands with ImageJ (https://imagej.nih.gov/ij/).
- Measure the intensity of each band in each lane with ImageJ.
- The relative editing efficiency is calculated by dividing the intensity of uncut band by the total intensities of all the bands.
Recipes
- Luria-Bertani medium
10 g/L tryptone
5 g/L yeast extract
10 g/L NaCl
Adjust pH to 7, autoclave at 121 °C for 20 min and store at room temperature - MgCl2 (1 M stock)
Dissolve 203.3 g of MgCl2·6H2O in 800 ml of deionized H2O
Adjust the volume to 1 L with deionized water - MES (0.5 M stock)
Dissolve 19.52 g MES in 200 ml deionized water
Adjust to pH 5.6 with 1 N KOH - Acetosyringone (1 M stock)
Dissolve 1.962 g acetosyringone in 10 ml of DMSO - Infiltration buffer
Note: The stock buffers above (Recipes 2-4) are filter sterilized with a 0.22-μm filter and stored at room temperature. The infiltration buffer is prepared freshly for each time.
10 mM MgCl2
10 mM MES
200 μM acetosyringone - 2x phosphate buffer (pH 7)
0.2 M NaH2PO4
0.2 M Na2HPO4 - First, make two solutions:
Solution A: dissolve 55.2 g NaH2PO4·H2O in 1 L deionized water
Solution B: dissolve 107.3 g Na2HPO4·7H2O in 1 L deionized water - Autoclave at 121 °C for 20 min and store them at room temperature
- To make 200 ml 2x phosphate buffer (pH 7), add 39.0 ml Solution A and 61.0 ml Solution B and then dilute with deionized water to 200 ml
- X-gluc substrate solution (Kabbage et al., 2011)
- Add 1 mg X-gluc in 0.1 ml methanol
- Then add 1 ml 2x phosphate buffer, 20 μl 0.1 M potassium ferricyanide, 10 μl 10% Triton X-100 and 850 μl deionized water
- PEG-MgCl2 solution
- Dissolve 30 g of PEG 8000 in 100 ml of 30 mM MgCl2
- Filter sterilize the solution with a 0.22-μm filter and store it at room temperature
Acknowledgments
The VIGE method is based on the report by Yin et al. (2015). The method of inoculating CaLCuV is based on Tang et al. (2010). The GUS staining assay is based on Kabbage et al. (2011). This work is supported by the National Basic Research Program of China (2014CB138400), the National Transgenic Program of China (2014ZX0800104B, 2014ZX08010-002 and 2014ZX08005-001) and the National Natural Science Foundation of China (31421001, 3142100007). We thank Alice L. Yu from University of North Carolina at Chapel Hill for proofreading the manuscript.
References
- Ali, Z., Abul-faraj, A., Li, L., Ghosh, N., Piatek, M., Mahjoub, A., Aouida, M., Piatek, A., Baltes, N. J., Voytas, D. F., Dinesh-Kumar, S. and Mahfouz, M. M. (2015). Efficient virus-mediated genome editing in plants using the CRISPR/Cas9 system. Mol Plant 8(8): 1288-1291.
- Aslanidis, C. and de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18(20): 6069-6074.
- Baltes, N. J., Gil-Humanes, J., Cermak, T., Atkins, P. A. and Voytas, D. F. (2014). DNA replicons for plant genome engineering. Plant Cell 26(1): 151-163.
- Höfgen, R. and Willmitzer, L. (1988). Storage of competent cells for Agrobacterium transformation. Nucleic Acids Res 16(20): 9877.
- Im, H. (2011). The inoue method for preparation and transformation of competent E. coli: “Ultra Competent” cells. Bio-protocol Bio101: e143.
- Kabbage, M., Ek-Ramos, M. and Dickman, M. (2011). A β-glucuronidase (GUS) based cell death assay. J Vis Exp 6(51).
- Lei, Y., Lu, L., Liu, H. Y., Li, S., Xing, F. and Chen, L. L. (2014). CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant 7(9): 1494-1496.
- Li, J. F., Zhang, D. and Sheen, J. (2015). Targeted plant genome editing via the CRISPR/Cas9 technology. Methods Mol Biol 1284: 239-255.
- Nekrasov, V., Staskawicz, B., Weigel, D., Jones, J. D. and Kamoun, S. (2013). Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31(8): 691-693.
- Paithankar, K. R. and Prasad, K. S. (1991). Precipitation of DNA by polyethylene glycol and ethanol. Nucleic Acids Res 19(6): 1346.
- Stemmer, W. P. and Morris, S. K. (1992). Enzymatic inverse PCR: a restriction site independent, single-fragment method for high-efficiency, site-directed mutagenesis. Biotechniques 13(2): 214-220.
- Tang, Y., Wang, F., Zhao, J., Xie, K., Hong, Y. and Liu, Y. (2010). Virus-based microRNA expression for gene functional analysis in plants. Plant Physiol 153(2): 632-641.
- Wang, H., La Russa, M. and Qi, L. S. (2016). CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem 85: 227-264.
- Yin, K., Han, T., Liu, G., Chen, T., Wang, Y., Yu, A. Y. and Liu, Y. (2015). A geminivirus-based guide RNA delivery system for CRISPR/Cas9 mediated plant genome editing. Sci Rep 5: 14926.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yin, K., Han, T. and Liu, Y. (2017). Use of Geminivirus for Delivery of CRISPR/Cas9 Components to Tobacco by Agro-infiltration. Bio-protocol 7(7): e2209. DOI: 10.21769/BioProtoc.2209.
Category
Plant Science > Plant transformation > Agrobacterium
Molecular Biology > DNA > Transformation
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.