- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Production, Purification and Crystallization of a Prokaryotic SLC26 Homolog for Structural Studies




Published: Vol 7, Iss 3, Feb 5, 2017 DOI: 10.21769/BioProtoc.2116 Views: 10089
Reviewed by: Arsalan DaudiChing Yao YangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2015
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The SLC26 or SulP proteins constitute a large family of anion transporters that are ubiquitously expressed in pro- and eukaryotes. In human, SLC26 proteins perform important roles in ion homeostasis and malfunctioning of selected members is associated with diseases. This protocol details the production and crystallization of a prokaryotic SLC26 homolog, termed SLC26Dg, from Deinococcus geothermalis. Following these instructions we obtained well-folded and homogenous material of the membrane protein SLC26Dg and the nanobody Nb5776 that enabled us to crystallize the complex and determine its structure (Geertsma et al., 2015). The procedure may be adapted to purify and crystallize other membrane protein complexes.
Keywords: Membrane transport proteinBackground
With few exceptions, structural characterization of membrane proteins involves challenges at the level of protein production, stabilization in the detergent-solubilized state, and crystallization. The strategy we have followed to overcome these hurdles relied on the efficient selection of SLC26 homologs with superior biochemical properties and the use of antibodies as crystallization chaperones (Geertsma et al., 2015). The procedures described here do not greatly deviate from those of colleagues, but on a few points we do follow alternative approaches. For example, for protein production we make use of the araBAD promoter (Guzman et al., 1995) and not the popular T7 promoter (Studier et al., 1990). In contrast to the T7 promoter, the PBAD promoter allows direct tuning of the protein production levels and its adjustment to the capacity of the downstream folding machinery, thereby reducing the formation of inclusion bodies (Geertsma et al., 2008). Furthermore, we prefer nanobodies, the variable domain of camelid heavy chain only antibodies (Pardon et al., 2014), as crystallization chaperones over the more commonly used Fabs. In our hands, the generation, selection, and production of nanobodies is far more robust and straightforward. Though we are aware that alternative protein production strategies (Henderson et al., 2000; Kunji et al., 2003; Miroux and Walker, 1996; Studier, 2005; Wagner et al., 2008) and crystallization chaperones (Koide, 2009; Seeger et al., 2013) exist, we did not explore these as the presented procedures proved very robust and successful.
Materials and Reagents
- Dialysis tube
MWCO 8 kDa (Carl Roth, catalog number: 1924.1 )
MWCO 3.5 kDa (Carl Roth, catalog number: E860.1 ) - Concentrators
MWCO 50 kDa (EMD Millipore, catalog number: UFC905024 )
MWCO 3 kDa (EMD Millipore, catalog number: UFC800324 ) - Crystallization plates (HAMPTON RESEARCH, catalog number: HR3-158 )
- Potter tube and piston (VWR, catalog numbers: 432-0205 and 432-0211 )
- 50 ml tube
- E. coli MC1061 (Coli Genetic Stock Center, catalog number: 6649 )
- Plasmid pBXNPHM3-Nb5776 (Figure 1A)
Note: This plasmid holds the gene coding for the nanobody under control of the araBAD promoter and results in the production of the nanobody fused to an N-terminal pelB leader sequence followed by decaHis-tag, MBP, and a HRV 3C protease site. It contains a beta-lactam antibiotic marker. Plasmid available for non-commercial use upon request. - Plasmid pBXC3GH-SLC26Dg (Figure 1B)
Note: This plasmid holds the gene coding for SLC26Dg under control of the araBAD promoter and results in the production of SLC26Dg fused to a C-terminal HRV 3C protease site, GFP, and a decaHis-tag. It contains a beta-lactam antibiotic marker. Plasmid available for non-commercial use upon request.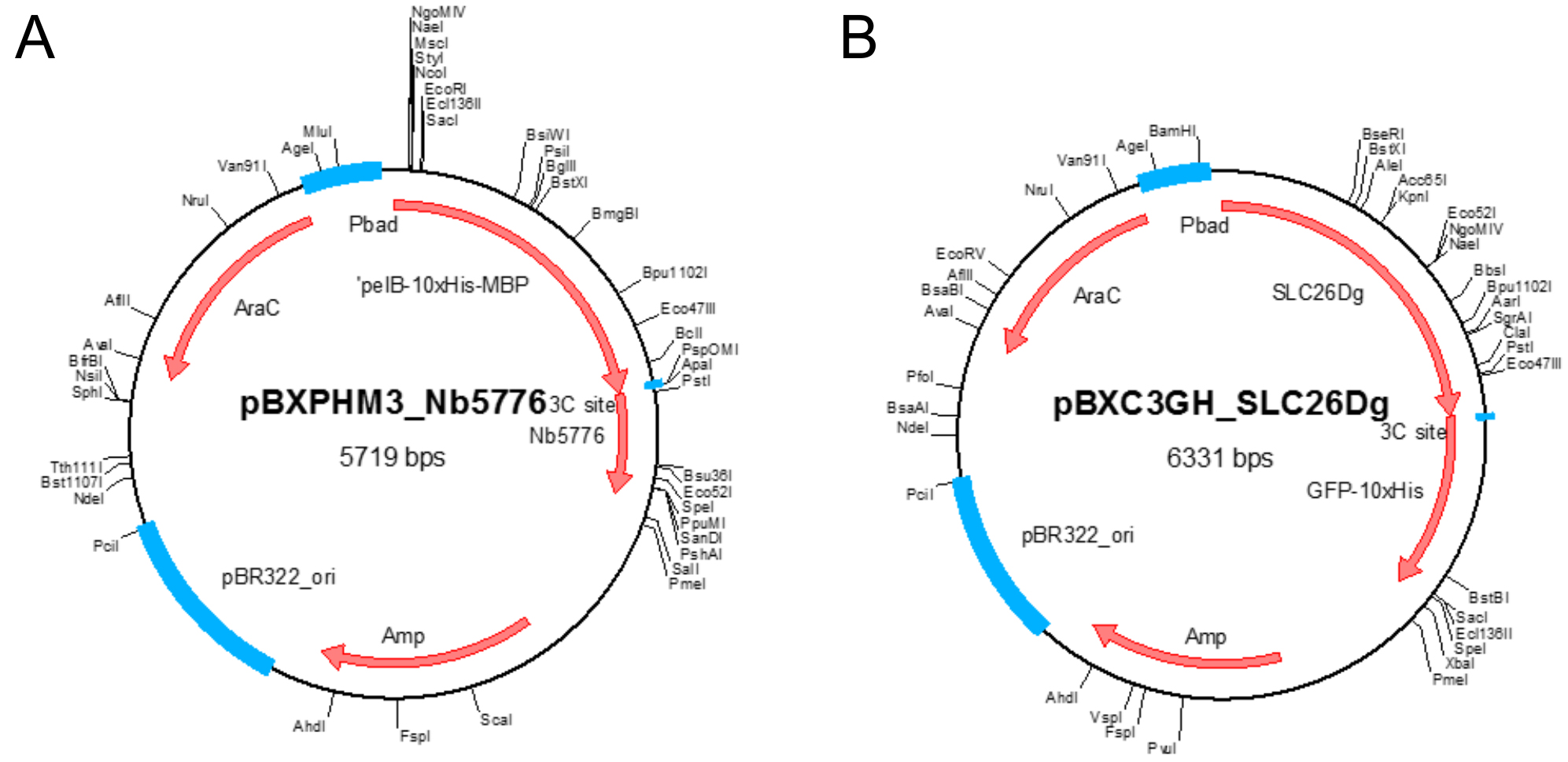
Figure 1. Plasmid maps. A. Plasmid map of pBXNPHM3-Nb5776; B. Plasmid map of pBXC3GH-SLC26Dg. Unique restriction sites and important features in the plasmids are indicated. - Ampicillin sodium salt (Carl Roth, catalog number: K029.2 )
- L-arabinose (20% w/v in ddH2O) (Carl Roth, catalog number: 5118.2 )
- 1 M KPi, pH 7.5 (see Recipes)
K2HPO4(AppliChem, catalog number: 121512 )
KH2PO4(AppliChem, catalog number: 131509 ) - Sodium chloride (NaCl, 2.5 M, ddH2O) (Carl Roth, catalog number: 3957.1 )
- Lysozyme (100 mg/ml in ddH2O) (AppliChem, catalog number: A3711 )
- DNAse I (2 mg/ml in ddH2O) (AppliChem, catalog number: A3778 )
- Magnesium sulphate heptahydrate (MgSO4, 1 M, ddH2O) (Carl Roth, catalog number: P027.2 )
- Phenylmethyl sulphonyl fluoride (PMSF, 200 mM in ethanol) (Carl Roth, catalog number: 6367.1 )
- NiNTA (50% w/v slurry in 20% EtOH) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 88223 )
- ddH2O
- Imidazole (2 M, pH 7.5, HCl, ddH2O) (Carl Roth, catalog number: X998.4 )
- HEPES (1 M, pH 7.5, NaOH, ddH2O) (Carl Roth, catalog number: HN78.2 )
- HRV 3C protease (homemade)
- Polypropylene glycol 2000 (10% v/v in ddH2O) (Sigma-Aldrich, catalog number: 81380-1L )
- EDTA, disodium salt dihydrate (Carl Roth, catalog number: X986.1 )
- Glycerol, 86% (w/v) (Carl Roth, catalog number: 4043.3 )
- Liquid nitrogen
- Decylmaltoside (10% w/v in ddH2O) (Anatrace, catalog numbers: D322S and D322LA )
- Ammonium formate (Carl Roth, catalog number: 5093.1 )
- Sodium acetate (Na-acetate) (Carl Roth, catalog number: 6773.2 )
- PEG400 (Sigma-Aldrich, catalog number: 91893 )
- Tryptone (AppliChem, catalog number: A1553 )
- Yeast extract (AppliChem, catalog number: A1552 )
- Agar (Applichem, catalog number: A0917 )
- LB agar (see Recipes)
- TB medium (see Recipes)
- Reservoir solution (see Recipes)
Equipment
- 5 L baffled flask
- Incubator at 37 °C
- Shaker with adjustable temperature (Infors, model: HT Multitron )
- Centrifuge for pelleting cultures (Thermo Fisher Scientific, Thermo Scientific, model: Thermo Sorvall Evolution RC )
- Homogenizer (IKA, model: ULTRA-TURRAX® T25 )
- High pressure cell disrupter (Avestin, model: Emulsiflex C3 )
- Nanodrop (Thermo Fisher Scientific, NanodropTM, model: 1000 , discontinued)
- Magnet stirrer (Heidolph Instruments, model: Hei-Mix S )
- SEC column Superdex 75 10/300 GL (GE Healthcare, catalog number: 17-5174-01 )
- SEC column Superdex 200 10/300 GL (GE Healthcare, catalog number: 28-9909-44 )
- Table-centrifuge (VWR, model: Micro Star 17 )
- HPLC (GE Healthcare, model: ÄKTAprime plus )
- Fermenter (BIOENGiNEERiNG, model: NLF 22, 30 L)
- Spectrophotometer (Amersham Biosciences, model: Ultrospec 10 )
Note: This product has been discontinued. - Ultracentrifuge (Beckman Coulter, model: Optima XPN-100K Ultracentrifuge )
Procedure
- Production and purification of Nb5776
- Transform E. coli MC1061 with pBXNPHM3-Nb5776. Plate on LB-agar plates containing 100 μg/ml ampicillin. Grow overnight at 37 °C.
- Pick a single colony to inoculate a preculture of 20 ml TB medium supplemented with 100 μg/ml ampicillin (TB/Amp). Grow overnight at 37 °C.
- Inoculate 1.5 L TB/Amp with 15 ml preculture in a 5 L baffled flask and cultivate at 37 °C. Adjust shaking speed to guarantee sufficient aeration while avoiding foam formation (~100 rpm).
- After 1.5 h set the temperature of the incubator to 25 °C and allow the culture to cool over the course of 1 h. Induce with a final concentration of 0.01% L-arabinose once the OD600 is between 0.5-1.0. Continue incubation overnight (~16 h).
- Harvest the cells by centrifugation at 5,500 x g for 15 min at 4 °C. From here on keep the sample cool (0-4 °C) and precool solutions and equipment beforehand.
- Resuspend the cells in ice-cold 50 mM KPi, pH 7.5, 150 mM NaCl to a final OD600 = 150-200.
- Add lysozyme (1 mg/ml final), DNAse I (20 μg/ml final concentration), and MgSO4 (1 mM final concentration).
- Homogenize the cells, e.g., using an Ultra-Turrax T25 homogenizer.
- Incubate for ~60 min at 4 °C while stirring.
- Disrupt cells by three passes at 10 kPsi using a high pressure cell disrupter (Emulsiflex, precooled).
- Add PMSF to a final concentration of 1 mM.
- Centrifuge the lysate for 30 min at 140,000 x g at 4 °C.
- Prepare the NiNTA column. Use 3 ml solid NiNTA. Wash the column with ~10 column volumes (CV; 30 ml) ddH2O and with ~10 CV (30 ml) 50 mM KPi, pH 7.5, 150 mM NaCl.
- Transfer the supernatant from step A12 to a new container and supplement with imidazole to a final concentration of 15 mM.
- Incubate the supernatant with NiNTA for 1 h at 4 °C, rotating.
- Drain column.
- Wash column with 20 CV (60 ml) 20 mM HEPES, pH 7.5, 500 mM NaCl, 50 mM imidazole.
- Elute column with 5x 0.5 CV (1.5 ml) 20 mM HEPES, pH 7.5, 150 mM NaCl, 300 mM imidazole.
- Determine protein concentration using a Nanodrop.
- Pool peak fractions and add 3C protease to a final protein:HRV 3C protease molar ratio of 5:1.
- Digest overnight at 4 °C while dialyzing against 20 mM HEPES, pH 7.5, 150 mM NaCl. Use a stirrer bar in the dialysis buffer. The MWCO of the dialysis tube should be 3.5 kDa. The dialysis volume should be sufficient to obtain a final imidazole concentration of 15 mM upon complete equilibration.
- Equilibrate the size-exclusion chromatography column (Superdex 75 10/300 GL) with 2.5 CV 10 mM HEPES, pH 7.5, 150 mM NaCl.
- Incubate the dialyzed material with 1 ml solid NiNTA (prewashed with ddH2O and dialysis buffer as in step A13) for 10 min at 4 °C, rotating.
- Collect the flow-through in a concentrator (3 kDa MWCO). The concentrator should be pre-run with ddH2O and flushed with dialysis buffer. Wash the NiNTA column with 3 CV dialysis buffer and collect this material in the concentrator as well.
- Centrifuge the concentrator for < 10 min at ~3,000 x g, 4 °C. Take it out and invert a few time to homogenize the solution (alternatively: pipet it up and down). Repeat until desired volume is reached (< 0.5 ml).
- Transfer sample to a precooled 1.5 ml cup. Centrifuge for 10 min at 16,000 x g in a table centrifuge at 4 °C.
- Transfer supernatant to a new precooled cup.
- Prepare the HPLC. Place new tubes in the fraction collector. Take the injection syringe apart and wash it with dialysis buffer. Remove bubbles from the syringe by rapidly pushing out the liquid. Pre-flush the needle and loop with dialysis buffer.
- Inject the sample into the loop. Start the run. Once the peak is approaching, change the fractionation volume to 0.3 ml. After the peak change to 1.4 ml fractionation.
- Determine which fractions contain the peak based on the chromatogram. Determine the protein concentration in these fractions using the Nanodrop.
- Pool peak fractions and concentrate (3 kDa MWCO) to a final concentration over 10 mg/ml.
- Store the sample at 4 °C until use. Nb5776 remains stable for at least 3 months.
- Transform E. coli MC1061 with pBXNPHM3-Nb5776. Plate on LB-agar plates containing 100 μg/ml ampicillin. Grow overnight at 37 °C.
- Production and purification of SLC26Dg
- Transform E. coli MC1061 with pBXC3GH_SLC26Dg. Plate on LB-agar plates containing 100 μg/ml ampicillin. Grow overnight at 37 °C.
- Pick a single colony to inoculate a preculture of 100 ml TB/Amp. Grow overnight at 37 °C.
- Inoculate 9 L TB/Amp with 90 ml preculture in a fermenter and cultivate at 37 °C. Adjust stirring and aeration to guarantee rapid growth. Adjust vessel pressure to approximately 1 bar overpressure to reduce foaming and supplement culture with small aliquots of a 10% (v/v) solution of polypropylene glycol 2000 if foam is formed. Continue cultivation until an OD600 of approximately 2 is reached.
- Gradually reduce the temperature to 25 °C over the course of 1 h. After an additional 20 min, induce the culture with 2.5 ml 20% (w/v) L-arabinose. Continue incubation overnight.
- Harvest the cells by centrifugation at 5,500 x g for 15 min at 4 °C. From here on keep the sample cool (0-4 °C) and precool solutions and equipment beforehand.
- Resuspend the cells in 50 mM KPi, pH 7.5, 150 mM NaCl to an OD600 = 150-200.
- Add lysozyme (1 mg/ml final), DNAse I (20 μg/ml final concentration), and MgSO4 (1 mM final concentration).
- Homogenize the cells, e.g., using an Ultra-Turrax T25 homogenizer.
- Incubate for ~60 min at 4 °C while stirring.
- Disrupt cells by three passes at 10 kPsi using a high pressure cell disrupter (Emulsiflex, precooled).
- Add PMSF (1 mM final concentration) and EDTA (5 mM final concentration).
- Perform a low spin centrifugation to remove unbroken cells, 15 min at 10,000 x g at 4 °C.
- Transfer supernatant to tubes suitable for ultracentrifugation.
- Perform a high spin centrifugation to pellet the membranes, 1 h at 160,000 x g at 4 °C.
- Determine the net-weight of the vesicle pellets.
- Resuspend the vesicles to ~1 g/2 ml in 50 mM KPi, pH 7.5, 150 mM NaCl, 10% glycerol using a Potter tube.
- Freeze the vesicles in liquid nitrogen in aliquots of 1/5/10 g.
- Store the aliquots at -80 °C until use. Vesicles can be stored under this condition over 1 year.
- Thaw 5 g membrane vesicles in a beaker with room temperature water and a stirrer bar. Keep the sample on ice once it is thawed.
- Equilibrate the decylmaltoside (sol-grade) powder to room temperature.
- Transfer the viscous vesicle suspension to a 50 ml tube. Wash the tube with ice-cold 50 mM KPi, pH 7.5, 150 mM NaCl, 10% glycerol and adjust the volume to 40 ml.
- Add imidazole to a final concentration of 15 mM.
- Add 0.625 g decylmaltoside to give a final concentration of ~1.6%. It is important to maintain a vesicles:detergent ratio of approximately 5 g:0.5-0.6 g in order to get good solubilization.
- Incubate 1 h at 4 °C, rotating.
- Centrifuge the material for 30 min at 160,000 x g at 4 °C.
- Prepare the NiNTA column. Use 3 ml solid NiNTA. Wash the column with ~10 CV (30 ml) ddH2O and with ~10 CV (30 ml) 50 mM KPi, pH 7.5, 150 mM NaCl, 10% glycerol.
- Incubate the supernatant with NiNTA for 1 h at 4 °C, rotating.
- Drain column.
- Wash column with 20 CV (60 ml) 20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 50 mM imidazole, 0.2% decylmaltoside.
- Elute column with five aliquots of 0.5 CV (1.5 ml) 20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 300 mM imidazole, 0.2% decylmaltoside.
- Determine protein concentration using the Nanodrop.
- Pool peak fractions and add 3C protease to a final protein:3C protease molar ratio of 5:1.
- Digest for 2 h at 4 °C while dialyzing against 20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 0.13% decylmaltoside. Use a stirrer bar in the dialysis buffer. The MWCO of the dialysis tube should be 8 kDa. The dialysis volume should be sufficient to obtain a final imidazole concentration of 15 mM upon complete equilibration.
- Equilibrate the size-exclusion chromatography column (Superdex 200 10/300 GL) with 2.5 CV 10 mM HEPES, pH 7.5, 150 mM NaCl, 0.2% decylmaltoside (highest purity).
- Incubate the dialyzed material with 0.6 ml solid NiNTA (prewashed with ddH2O and dialysis buffer as in step B26) for 10 min at 4 °C, rotating.
- Collect the flow-through in a concentrator (50 kDa MWCO). The concentrator should be pre-run with ddH2O and flushed with dialysis buffer. Wash the NiNTA column with 3 CV dialysis buffer and collect this flow-through in the concentrator as well.
- Centrifuge the concentrator for < 10 min at 3,000 x g, 4 °C. Take it out and invert a few time to homogenize the solution (alternatively: pipet it up and down). Repeat until desired volume is reached (< 0.5 ml).
- Transfer sample to a precooled 1.5 ml cup. Centrifuge for 10 min at > 16,000 x g in a table centrifuge at 4 °C.
- Transfer supernatant to a new precooled cup.
- Prepare the HPLC. Place new tubes in the fraction collector. Take the injection syringe apart and wash it with dialysis buffer. Remove bubbles from the syringe by rapidly pushing out the liquid. Pre-flush the needle and loop with dialysis buffer.
- Inject the sample into the loop. Start the run. Once the peak is approaching, change the fractionation volume to 0.3 ml. After the peak change to 1.4 ml fractionation.
- Determine which fractions contain the peak based on the chromatogram. Determine the protein concentration in these fractions using the Nanodrop.
- Pool peak fractions and concentrate (50 kDa MWCO) to a final concentration over 10 mg/ml.
- Do not store the sample but immediately proceed with complex formation and crystallization.
- Transform E. coli MC1061 with pBXC3GH_SLC26Dg. Plate on LB-agar plates containing 100 μg/ml ampicillin. Grow overnight at 37 °C.
- Complex formation and crystallization of SLC26Dg-Nb5776
- Supplement purified and concentrated Nb5776 with decylmaltoside (10% w/v stock) to a final concentration of 0.2% (w/v).
- Mix purified SLC26Dg and Nb5776 at a molar ratio of 1:2. Incubate for 10 min at 4 °C.
- Submit the sample to SEC using a Superdex 200 10/300 GL column equilibrated with 10 mM HEPES, pH 7.5, 150 mM NaCl, 0.2% decylmaltoside.
- Determine which fractions contain the peak based on the chromatogram. Determine the protein concentration in these fractions using the Nanodrop. Verify the presence of both proteins in the fractions by SDS-PAGE.
- Pool peak fractions and concentrate (50 kDa MWCO) to a final concentration of approximately 9-12 mg/ml.
- Grow SLC26Dg-Nb5776 crystals in sitting drops (1 μl protein + 1 μl reservoir solution [see Recipes]) by vapor diffusion at 4 °C. Mix protein and reservoir solutions in a 1:1 ratio. Crystals appear after approximately 1 week.
- Supplement purified and concentrated Nb5776 with decylmaltoside (10% w/v stock) to a final concentration of 0.2% (w/v).
Data analysis
Representative data is depicted in Figures 2 and 3.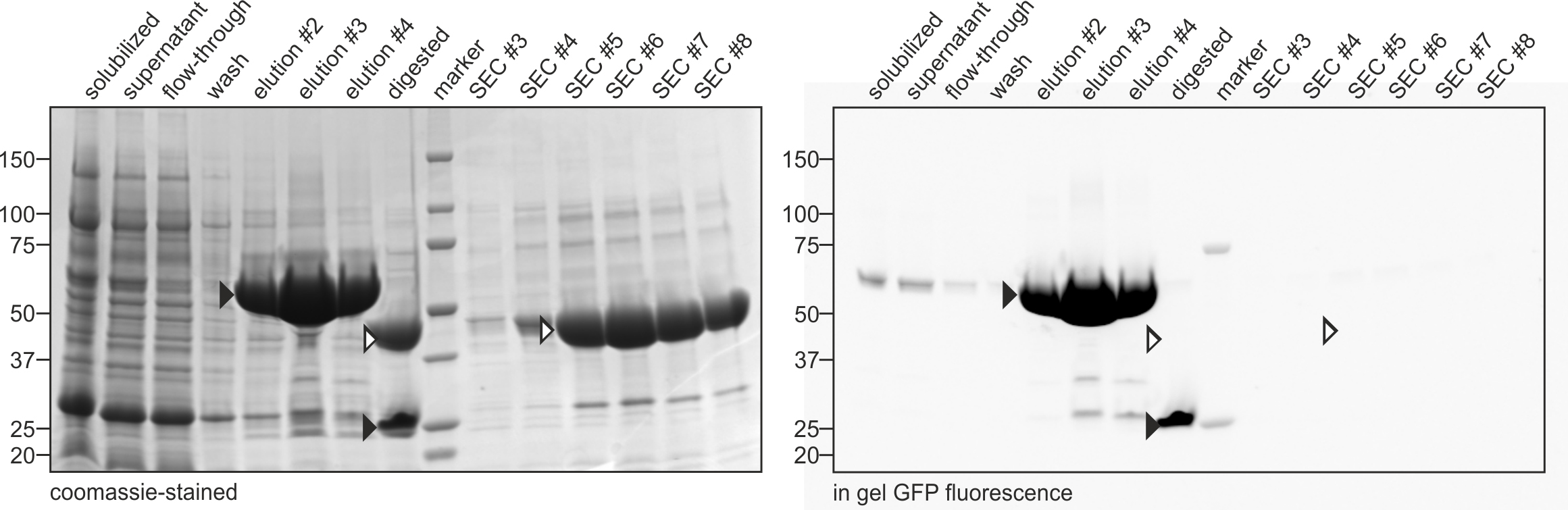
Figure 2. Purification of SLC26Dg. SDS-PAGE analysis of samples obtained at different steps during the purification of SLC26Dg according to the protocol detailed here. ‘Elution’, ‘digested’, and ‘SEC’, refer to samples obtained after elution from the NiNTA column, after digestion with HRV 3C protease, and after elution from the SEC column, respectively. Black and white arrows indicate bands containing GFP (fused to SLC26Dg or alone) and SLC26Dg, respectively. The right panel represents an image of the SDS-PAGE gel obtained by measuring the in gel GFP fluorescence. The left panel represents a coomassie-stained image of the same gel. Molecular weight markers (in kDa) are indicated on the left.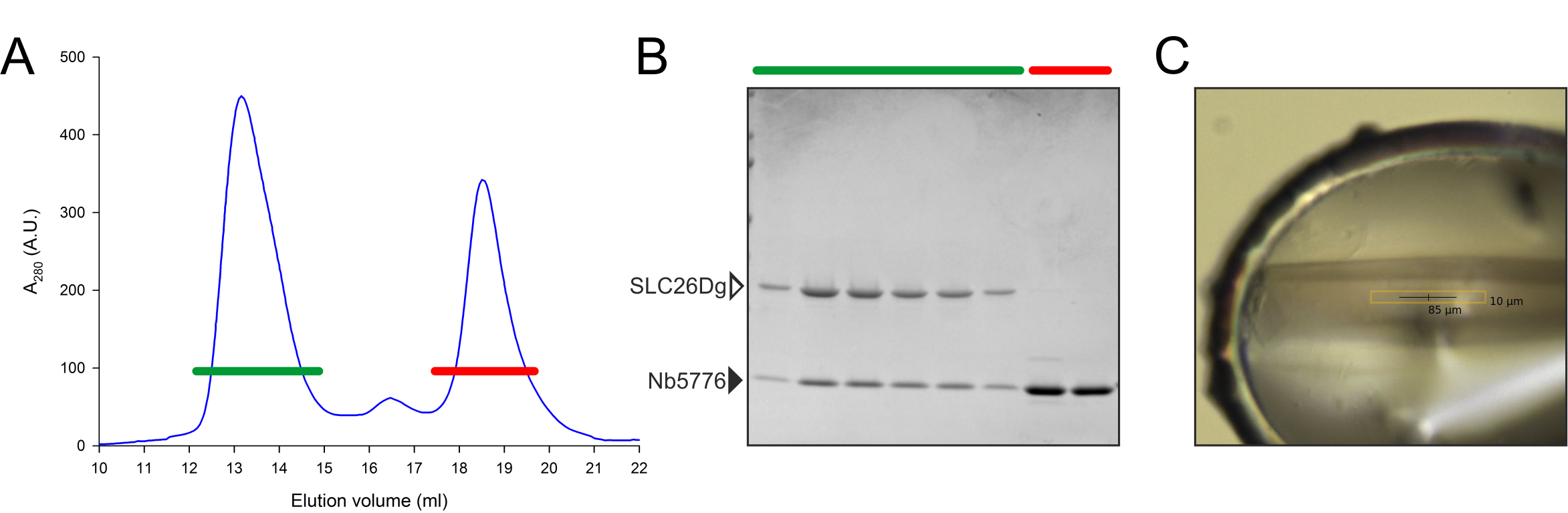
Figure 3. Complex formation of SLC26Dg and Nb5776. A. Size-exclusion chromatogram of SLC26Dg pre-incubated with an excess of Nb5776 as indicated in the protocol. B. SDS-PAGE analysis of the relevant peak fractions from the SEC column. C. Protein crystal of the SLC26Dg-Nb5776 complex.
Recipes
- LB agar (1 L)
10 g tryptone
5 g yeast extract
5 g NaCl
15 g agar
Add demineralized H2O to 1 L and autoclave at 121 °C for 20 min - TB medium (1 L)
- 12 g tryptone
24 g yeast extract
8.6 ml 86% (w/v) glycerol
Add demineralized H2O up to 950 ml - 50 ml 1.44 M K2HPO4, 0.34 M KH2PO4 in ddH2O
Autoclave both solutions separately (at 121 °C for 20 min) and mix before use - 1 M KPi, pH 7.5 (200 ml)
Prepare a 200 ml solution of 1 M K2HPO4 and a 100 ml solution of 1 M KH2PO4 using ddH2O
Mix both solutions in an approximate ratio of 4:1 (K2HPO4:KH2PO4) under constant stirring while measuring the pH
Set the pH by adjusting with the basic (K2HPO4) or acidic (KH2PO4) component - Reservoir solution
1 M ammonium formate, 50 mM Na-acetate, pH 4.5
45-50% PEG400 (w/v)
Acknowledgments
The protocols detailed here were developed in the laboratory of Prof. Raimund Dutzler at the University of Zurich and benefitted from helpful suggestions from Drs. Sandra Markovic, Ricarda J.C. Hilf, Ines Ehrnstorfer, Stefan Warmuth, Iwan Zimmermann, and Prof. Markus Seeger. Prof. Jan Steyaert and Dr. Els Pardon from the Vrije Universiteit Brussel are acknowledged for the generation and selection of Nb5776. Beat Blattman and Céline Stutz-Ducommun from the Protein Crystallization Center of the University of Zürich are acknowledged for their support in establishing crystallization conditions for SLC26Dg/Nb5776. E.R.G. acknowledges support by a long-term fellowship from the Human Frontier Science Program (LT-00899/2008) and the German Research Foundation through the Cluster of Excellence Frankfurt ‘Macromolecular Complexes’.
References
- Geertsma, E. R., Chang, Y. N., Shaik, F. R., Neldner, Y., Pardon, E., Steyaert, J. and Dutzler, R. (2015). Structure of a prokaryotic fumarate transporter reveals the architecture of the SLC26 family. Nat Struct Mol Biol 22(10): 803-808.
- Geertsma, E. R., Groeneveld, M., Slotboom, D. J. and Poolman, B. (2008). Quality control of overexpressed membrane proteins. Proc Natl Acad Sci U S A 105(15): 5722-5727.
- Guzman, L. M., Belin, D., Carson, M. J. and Beckwith, J. (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177(14): 4121-4130.
- Henderson, P. J., Hoyle, C. K. and Ward, A. (2000). Expression, purification and properties of multidrug efflux proteins. Biochem Soc Trans 28(4): 513-517.
- Koide, S. (2009). Engineering of recombinant crystallization chaperones. Curr Opin Struct Biol 19(4): 449-457.
- Kunji, E. R., Slotboom, D. J. and Poolman, B. (2003). Lactococcus lactis as host for overproduction of functional membrane proteins. Biochim Biophys Acta 1610: 97-108.
- Miroux, B. and Walker, J. E. (1996). Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260(3): 289-298.
- Pardon, E., Laeremans, T., Triest, S., Rasmussen, S. G., Wohlkonig, A., Ruf, A., Muyldermans, S., Hol, W. G., Kobilka, B. K. and Steyaert, J. (2014). A general protocol for the generation of Nanobodies for structural biology. Nat Protoc 9(3): 674-693.
- Seeger, M. A., Zbinden, R., Flutsch, A., Gutte, P. G., Engeler, S., Roschitzki-Voser, H. and Grutter, M. G. (2013). Design, construction, and characterization of a second-generation DARPin library with reduced hydrophobicity. Protein Sci 22(9): 1239-1257.
- Studier, F. W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41(1): 207-234.
- Studier, F. W., Rosenberg, A. H., Dunn, J. J. and Dubendorff, J. W. (1990). Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol 185: 60-89.
- Wagner, S., Klepsch, M. M., Schlegel, S., Appel, A., Draheim, R., Tarry, M., Hogbom, M., van Wijk, K. J., Slotboom, D. J., Persson, J. O. and de Gier, J. W. (2008). Tuning Escherichia coli for membrane protein overexpression. Proc Natl Acad Sci U S A 105(38): 14371-14376.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chang, Y., Shaik, F. R., Neldner, Y. and Geertsma, E. R. (2017). Production, Purification and Crystallization of a Prokaryotic SLC26 Homolog for Structural Studies. Bio-protocol 7(3): e2116. DOI: 10.21769/BioProtoc.2116.
Category
Microbiology > Microbial biochemistry > Protein > Isolation and purification
Biochemistry > Protein > Expression
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.