- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Assessment of Cellular Redox State Using NAD(P)H Fluorescence Intensity and Lifetime



Published: Vol 7, Iss 2, Jan 20, 2017 DOI: 10.21769/BioProtoc.2105 Views: 14917
Reviewed by: Nicoletta CordaniRakesh BamAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
NADH and NADPH are redox cofactors, primarily involved in catabolic and anabolic metabolic processes respectively. In addition, NADPH plays an important role in cellular antioxidant defence. In live cells and tissues, the intensity of their spectrally-identical autofluorescence, termed NAD(P)H, can be used to probe the mitochondrial redox state, while their distinct enzyme-binding characteristics can be used to separate their relative contributions to the total NAD(P)H intensity using fluorescence lifetime imaging microscopy (FLIM). These protocols allow differences in metabolism to be detected between cell types and altered physiological and pathological states.
Keywords: NADHBackground
The reduced form of the redox cofactor nicotinamide adenine dinucleotide (NADH) and its phosphorylated counterpart NADPH are intrinsically fluorescent, both absorbing light at wavelengths of 340 (± 30) nm and emitting at 460 (± 50) nm (Patterson et al., 2000). These spectral characteristics are lost upon oxidation to NAD+ or NADP+ (De Ruyck et al., 2007). The redox balances of the separate NAD and NADP pools dictate contrasting metabolic processes (Ying, 2008), as shown in Figure 1. NAD acts as an electron acceptor for the oxidation of sugar, lipid and amino acid substrates in the mitochondria by the tricarboxylic acid (TCA) cycle and as an electron donor to the electron transport chain (ETC) on the inner mitochondrial membrane (IMM), fuelling the pumping of protons into the intermembrane space to act as a power source for the synthesis of adenosine triphosphate (ATP) by the F1FO ATP synthase (Osellame et al., 2012). The balance of NADH to NAD+ in the mitochondria therefore reflects the balance of TCA cycle to ETC activity. ETC dysfunction causes increases in the NADH/NAD+ ratio and the production of potentially damaging reactive oxygen species (ROS) (Murphy, 2009). The cell’s antioxidant defences require the NADP pool to provide reducing equivalents for their maintenance, so the NADPH/NADP+ ratio must be maintained high (> 3) (Pollak et al., 2007). The redox state of the mitochondrial NAD pool and the relative abundance of NADPH are therefore key factors in the level of oxidative stress in a cell type.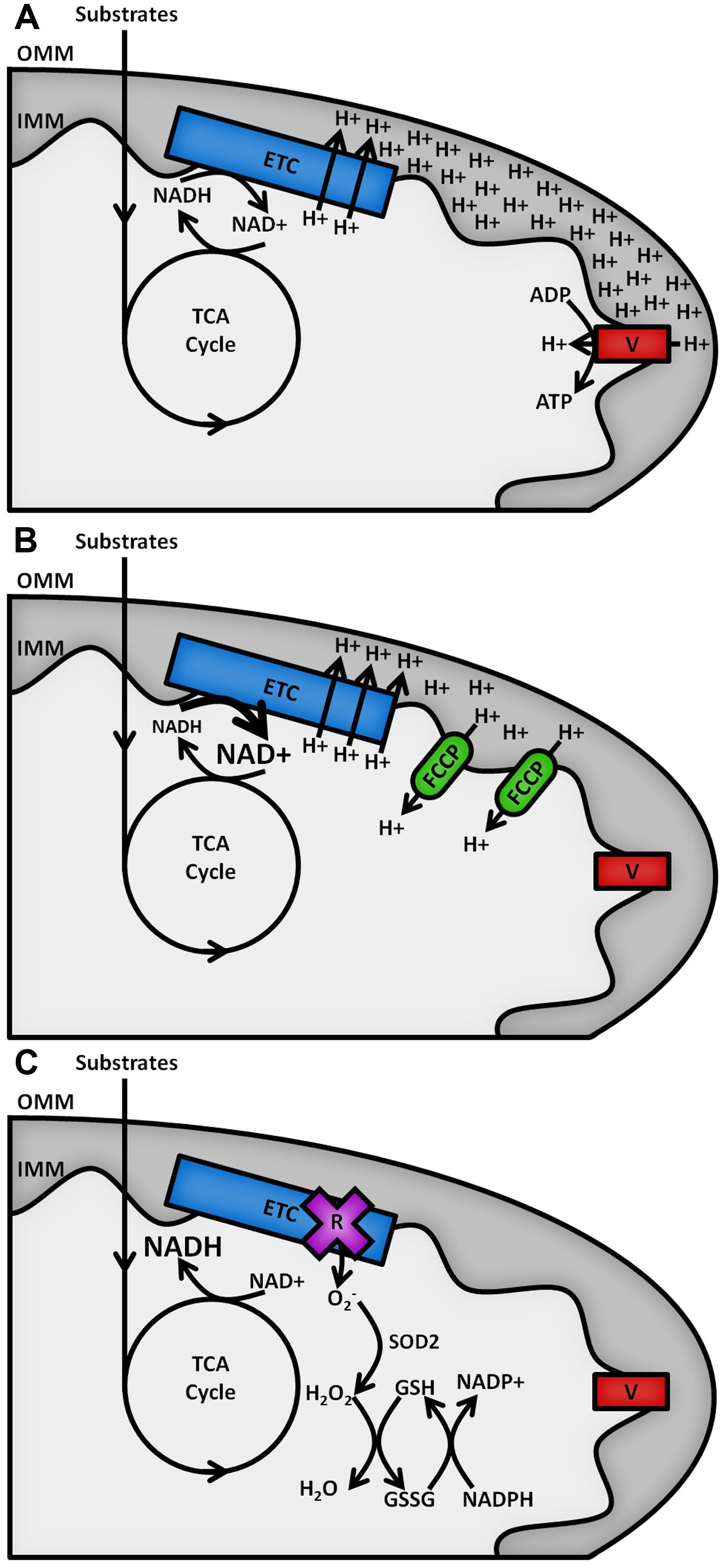
Figure 1. Schematic outline of mitochondrial NAD(P)H metabolism. Substrate oxidation in the TCA cycle passes electrons to NAD+, forming NADH. A. Under resting conditions, electrons carried by NADH are passed along the ETC, powering the pumping of protons from the mitochondrial matrix across the inner mitochondrial membrane (IMM) into the intermembrane space. The resulting proton gradient powers the production of ATP at complex V of the ETC (F1FO ATP synthase). B. Addition of an uncoupler such as carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) allows protons to leak back across the IMM, causing the rate of oxidation of NADH at the ETC to increase to restore the membrane gradient. C. Inhibition of the ETC by rotenone halts NADH oxidation and causes an increase in the production of superoxide (O2-), the proximal source of mitochondrial ROS. This is neutralised upon its conversion into water by the superoxide dismutase (SOD2) and glutathione (GSH/GSSG) antioxidant defence systems, maintained by NADPH.
Here, we describe protocols for the assessment of the mitochondrial NADH/NAD+ ratio and the NADPH/NADH balance that rely on the fluorescence of these cofactors when reduced. Their identical absorption and emission spectra leads the combined signal to be termed NAD(P)H (Blacker and Duchen, 2016). Measuring the change in NAD(P)H fluorescence using a confocal microscope following the application of an ETC uncoupler and inhibitor allows the mitochondrial NADH/NAD+ balance to be estimated (Duchen et al., 2003). To discriminate between the relative contributions of NADH and NADPH to the total signal, fluorescence lifetime imaging microscopy (FLIM) must be introduced (Blacker et al., 2014). These protocols describe, in further detail, methods used in Tosatto et al. (2016) to investigate the role of the selective channel responsible for mitochondrial calcium uptake, the mitochondrial calcium uniporter (MCU), in the progression of breast cancer. Basic understanding of confocal microscopy is assumed. For background, readers are directed to Pawley et al. (2012).
Materials and Reagents
- NuncTM cell culture treated EasYFlasksTM (75 cm2, filter closure) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 156499 )
- Falcon tubes (15 ml) (VWR, catalog number: 734-0451 )
- Tips
- Falcon tubes (50 ml) (VWR, catalog number: 734-0448 )
- Eppendorf tubes (1.5 ml) (VWR, catalog number: 211-2520 )
- NuncTM cell-culture treated multidishes (6 wells) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 140675 )
- Coverslips (22 mm, thickness No.1) (VWR, catalog number: 631-0158 )
- Syringe (20 ml, VWR, catalog number: 613-3921 )
- Filter steriliser (0.2 μm, VWR, catalog number: 514-0064 )
- MDA-MB-231 cells (ATCC, catalog number: HTB-26 )
- Advanced Dulbecco’s modified Eagle’s medium (DMEM) (500 ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 12491023 )
- Fetal bovine serum (FBS, heat inactivated, 50 ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 10500064 )
- GlutaMAXTM (5 ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 35050061 )
- Penicillin-streptomycin (10,000 U ml-1, 5 ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Trypsin-EDTA (0.25%, with phenol red, 100 ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 25200056 )
- FCCP (10 mg) (Sigma-Aldrich, catalog number: C2920 )
- Rotenone (5 g) (Sigma-Aldrich, catalog number: R8875 )
- Ethanol (1 L) (Sigma-Aldrich, catalog number: 02860 )
- Dulbecco’s modified Eagle’s medium powder (without glucose, L-glutamine, phenol red, sodium pyruvate and sodium bicarbonate) (Sigma-Aldrich, catalog number: D5030 )
- D-(+) glucose (100 g) (Sigma-Aldrich, catalog number: G8270 )
- Sodium pyruvate (25 g) (Sigma-Aldrich, catalog number: P2256 )
- HEPES (100 g) (Sigma-Aldrich, catalog number: H3375 )
- Sodium hydroxide solution (1 N, 1 L) (Sigma-Aldrich, catalog number: 71463 )
- Hydrochloric acid solution (1 N, 1 L) (Sigma-Aldrich, catalog number: 71763 )
- Routine cell culture medium (500 ml) (see Recipes)
- Live-cell imaging medium (50 ml) (see Recipes)
- ETC perturbations (see Recipes)
Equipment
- Incubator (37 °C, 5% CO2) (Thermo Fisher Scientific, Thermo ScientificTM, model: HERAcellTM 150i )
- Centrifuge with 15 ml Falcon tube capacity (Mega Star 1.6) (VWR, catalog number: 521-1749 )
- Haemocytometer (Neubauer) (VWR, catalog number: 630-1509 )
- Laser-scanning microscope (Zeiss, model: LSM510 )
- 40x magnification objective
- Blackout curtains, for room and covering FLIM microscope (see Figure 2)

Figure 2. Equipment for high signal to noise FLIM. A. Zeiss LSM510 with Coherent Chameleon for two-photon excitation and Becker & Hickl HPM-100-40 detector and SPC830 counting electronics. B. Blackout curtains surrounding both the microscope room and the microscope itself act to ensure that background noise in the FLIM images is kept to a minimum. - Emission filters for NAD(P)H fluorescence (435-485 nm)
- For single-photon excitation:
- 351 nm laser source (Enterprise UV, Coherent)
- Long-pass dichroic (375 nm cutoff)
- Quartz microscope optics
- For two-photon excitation:
- Ti:sapphire laser modelocked at 720 nm (Chameleon Ultra, Coherent)
- Short pass dichroic (650 nm cutoff)
- Non-descanned detection
- For FLIM:
- Detector with single photon sensitivity (Becker & Hickl, model: HPM-100-40)
- Time-correlated single photon counting (TCSPC) electronics (Becker & Hickl, model: SPC-830)
- Pulsed excitation source (two-photon excitation by Ti:sapphire laser at 720 nm; Chameleon Ultra, Coherent)
- Heated microscope stage (custom built to hold imaging rings, see Figure 3)
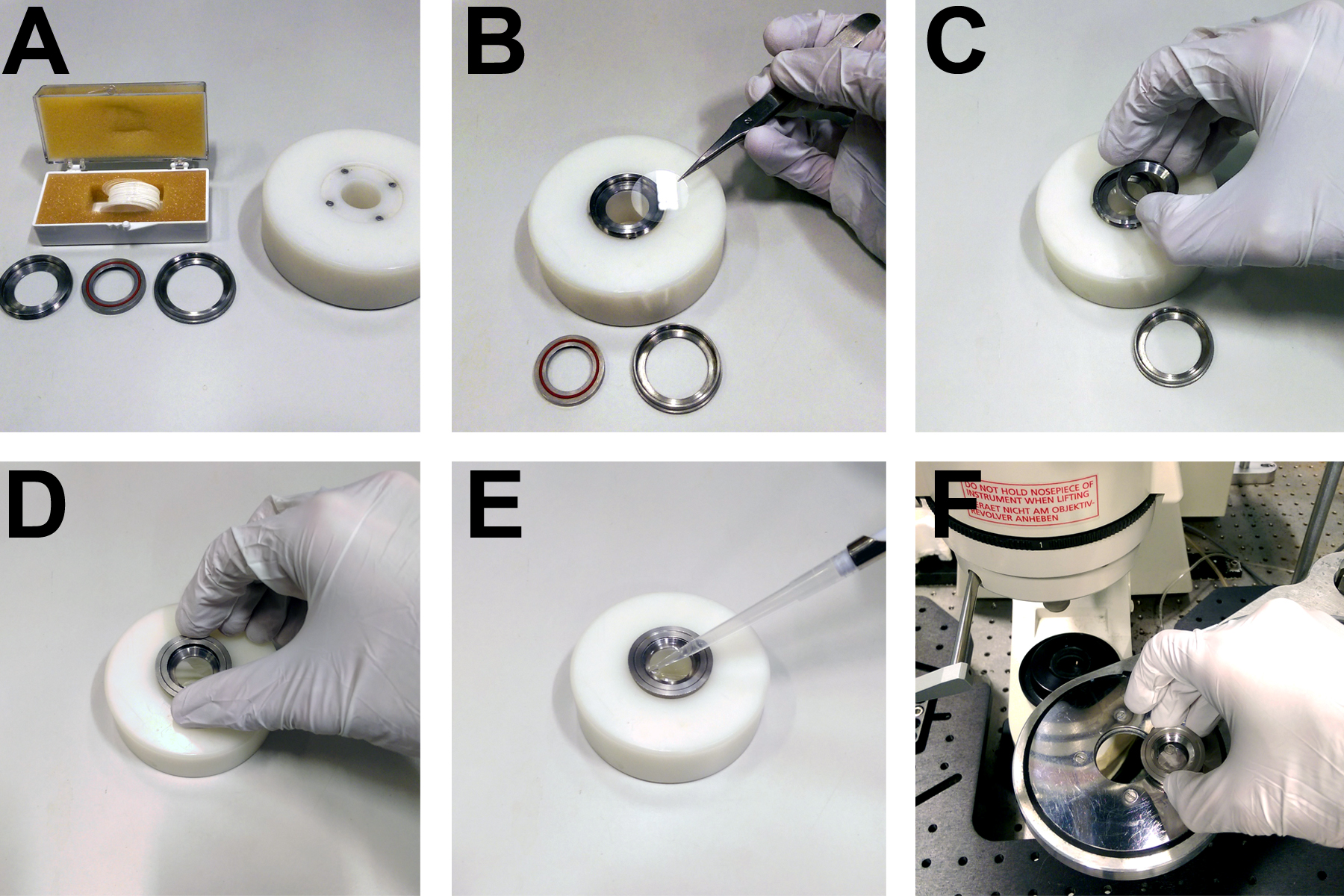
Figure 3. Apparatus for mounting coverslips on the microscope. A. Custom built rings and mounting block for securing 22 mm circular coverslips. B. Coverslips are placed directly onto the base of a metal ring. C. A concentric ring with a rubber seal is placed on top of the coverslip. D. A third ring is screwed onto the first two in order to form a watertight seal over the coverslip, allowing (E) the imaging buffer to be pipetted onto the cells. F. The imaging ring and cells then fit into a heated microscope stage.
- Straight-tip forceps (VWR, catalog number: 232-0094 )
- Imaging rings (custom built, see Figure 3)
- Inverted, phase-contrast, bench top microscope (VWR, catalog number: 630-2145 ; ZEISS, model: Primovert )
- Laboratory balance (0.1 mg resolution) (Mettler-Toledo, catalog number: 30029067 )
- pH meter (Mettler-Toledo International, catalog number: 30266626 )
- Tally counter (VWR, catalog number: 720-1984 )
Software
- Microscope control software (Zeiss ZEN)
- FLIM acquisition software (Becker & Hickl SPCM)
- FLIM fitting software (Becker & Hickl SPCImage)
- Image analysis software (NIH ImageJ)
Procedure
- Routine culture of MDA-MB-231 cells
- Culture control (scrambled plasmid) and MCU knockdown MDA-MB-231 cells in Advanced DMEM supplemented with 10% FBS, 2 mM GlutaMAXTM, 100 U ml-1 penicillin and 100 μg ml-1 streptomycin.
- Maintain cells as monolayers in 75 cm2 tissue culture flasks containing 10-12 ml growth medium in a 37 °C, 5% CO2 incubator.
- Passage every 3-4 days is ensured by splitting at a 1:10 ratio at 70-80% confluence.
- Dissociate cells by an initial wash with 2 ml trypsin followed by incubation with 2 ml fresh trypsin for 5 min.
- Plating of cells for microscopy
- Discard growth media from 75 cm2 flask and detach cells by incubating with 3 ml trypsin for 5 min.
- Collect cells and inhibit the enzyme by adding 5 ml growth medium.
- Transfer the contents of the flask to a 15 ml Falcon tube and centrifuge at 400 x g for 5 min at room temperature (24 °C).
- Resuspend cells in 5 ml fresh medium and transfer a small amount (~100 μl) to an Eppendorf tube for cell counting.
- Count cell concentration in resuspension using haemocytometer and tally counter on bench top microscope, ensuring to mix contents of the Eppendorf tube thoroughly with pipette to account for settling.
- Using results of cell counting, pipette 1,800,000 cells from cell suspension into a new 50 ml Falcon tube (300,000 cells per well of a 6 well plate). Top up tube to contain a total of 12 ml (2 ml total medium per well).
- Using forceps, transfer one coverslip into each well of a 6 well plate.
- Pipette 2 ml of the 1,800,000 cell mix into each well, again ensuring to mix contents of the tube using pipette prior to transfer.
- Place 6 well plate in incubator and leave overnight to allow attachment of cells to coverslips prior to imaging the next day.
- Repeat process for each cell type under analysis in a separate 6 well plate.
- NAD redox state assay
- Use forceps to transfer a coverslip from the 6 well plate into the metal imaging ring (see Figure 3). Tighten until a small amount of resistance is felt to ensure a watertight seal.
- Wash cells by gently (to ensure cells are not detached) pipetting 400 μl of imaging buffer onto coverslip in order to remove remaining phenol red.
- Dispose washing medium and gently pipette 400 μl of imaging buffer onto cells.
- Transfer coverslip onto heated stage of confocal microscope and bring cells into focus under brightfield illumination.
- With 375 nm long pass dichroic and 435-485 nm emission filters in place, gradually increase intensity of 351 nm excitation laser until NAD(P)H fluorescence is clearly observed.
- Search coverslip and magnify image such that 10-20 cells can be observed in the field of view.
- Begin a time-series acquisition, imaging the field of view every 2 min.
- After 4 images (6 min from the start of time-series), gently add 100 μl of 5 μM FCCP solution directly onto cells, giving a final FCCP concentration of 1 μM. The NAD(P)H fluorescence signal should begin to decrease due to increased ETC activity.
- Following acquisition of 4 images under FCCP treatment (8 min after addition), gently add 100 μl of 30 μM rotenone solution directly onto cells, giving a final rotenone concentration of 5 μM. The NAD(P)H fluorescence signal should begin to increase, due to inhibition of the ETC.
- Terminate experiment 10 min after rotenone addition, giving a total experiment time of 24 min.
- Dispose of cells and use 100% ethanol to clean any experimental equipment that may come into contact with FCCP or rotenone (e.g., imaging rings, microscope objective).
- Repeat procedure for all coverslips and save data for subsequent analysis.
- NAD(P)H FLIM assay
- Prepare a coverslip in the metal imaging ring with 400 μl imaging buffer (identically as for the redox state assay) and mount on microscope, adjusting focus to observe cells clearly under brightfield illumination.
- With 650 nm short pass dichroic and 435-485 nm emission filters in place, gradually increase intensity of 720 nm excitation laser until NAD(P)H fluorescence can be observed with the internal detectors of the microscope.
- Search coverslip and magnify image such that 10-20 cells can be observed in the field of view.
- Adjust microscope settings to send fluorescence signal to non-descanned port where FLIM detector is located.
- Begin FLIM data acquisition, scanning for 2 min total.
- Move to a new location on the coverslip and repeat process. Acquire 3-5 images per coverslip.
- Dispose of cells and use 100% ethanol to clean any experimental equipment that may come into contact with FCCP or rotenone (e.g., imaging rings, microscope objective).
- Repeat procedure for all coverslips and cell types.
Data analysis
- NAD redox state assay (see Figure 4)
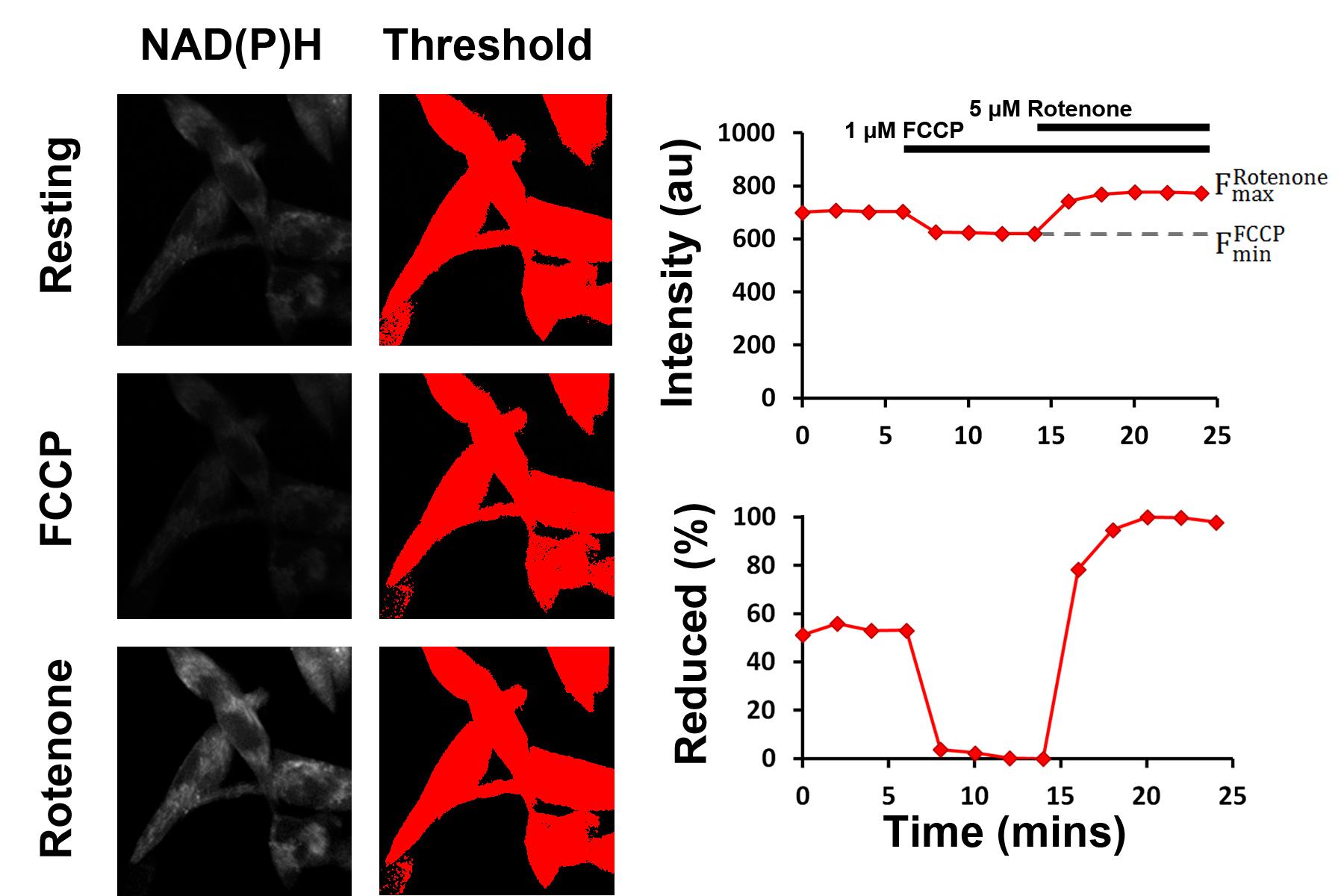
Figure 4. Schematic representation of the mitochondrial NAD redox state assay. The fluorescence intensities following treatment with 1 μM FCCP and 1 μM rotenone are used as the dynamic range through which the resting fluorescence intensity, and thereby the resting percentage of reduced NADH, can be assessed. Thresholding is used to select pixels above a defined brightness value (i.e., pixels containing cells), reducing the impact of background noise. - Import time series images into ImageJ. Dataset will be represented as a ‘stack’, with the x- and y-axes representing the spatial coordinates of the images and the z-axis representing the time since the start of the experiment. The time series can be navigated by moving the horizontal scroll bar.
- Use the threshold tool to highlight cell regions in red. The minimum and maximum values should be chosen such that no background signal is selected at any of the time points, and no bright intracellular regions (e.g., with rotenone) should be ignored.
- Ensure that measurements are limited to thresholded regions, negating the impact of background fluorescence, by clicking ‘Limit to threshold’ in the ‘Set Measurements’ dialog.
- Plot the time evolution of the NAD(P)H fluorescence signal by selecting Plot Z-axis Profile from the Stacks menu. Export the data to Microsoft Excel.
- Repeat this process for each time-series acquired.
- In Excel, scale the intensities at each time point F(t) as a percentage of the minimum (with FCCP) and maximum (with rotenone) values of its time-series by applying the following formula:
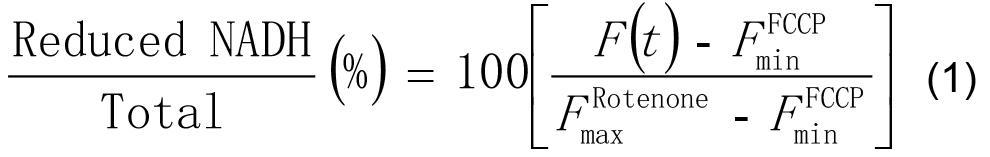
- Correct application of this procedure will leave the minimum value of each time-series at 0% and the maximum value as 100%.
- For each cell line, take a mean of all scaled data points acquired under resting conditions to calculate the final Reduced NADH (%) value. Express the uncertainty as the standard error of these points. Statistical significance of any differences between the Reduced NADH (%) values for each cell line can be assessed using a Wilcoxon signed-rank test.
- NAD(P)H FLIM assay (see Figure 5)
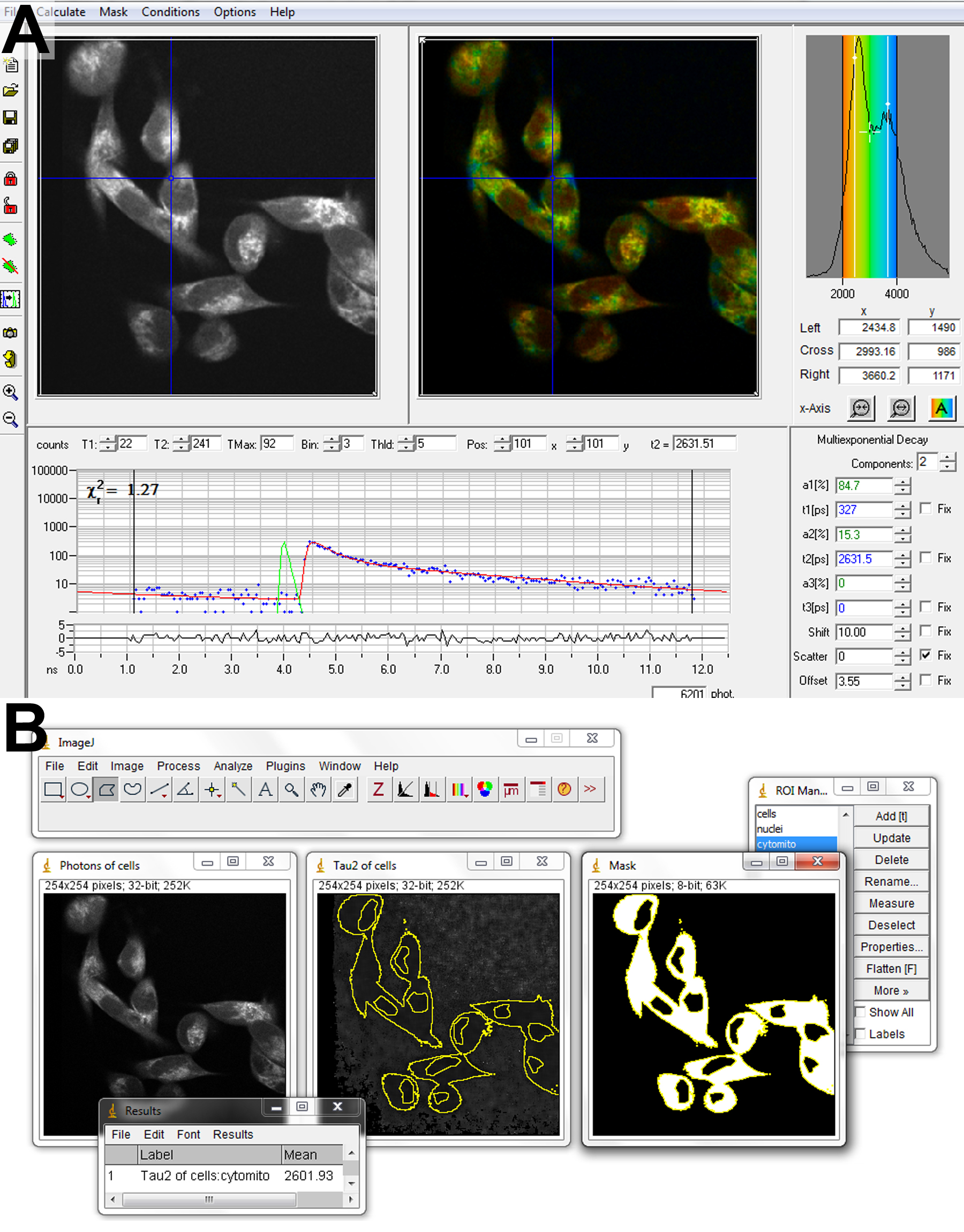
Figure 5. Software for FLIM analysis. Fitting of biexponential fluorescence decays to a FLIM image is performed in SPCImage (A). Parameter matrices are exported to ImageJ (B) for subsequent analysis. Regions of interest containing cytosol and mitochondria are created by combining a mask of the entire cell, generated by thresholding, and excluding the nuclear regions, drawn by hand. - In SPCImage, import the FLIM image and adjust the settings to fit 2 decay components at each pixel.
- To ensure fits of sufficient accuracy, binning should be increased until more than 100 counts are contained in the peak of the decay in the dimmest cytosolic pixel of interest (Blacker et al., 2014). For 2 min acquisitions with laser powers kept sufficiently low to avoid cell damage, this typically requires a binning factor of 2 to 4, corresponding to the summation of data from between 24 and 80 surrounding pixels.
- Begin the fitting procedure at each pixel by choosing Calculate Decay Matrix. This process could take up to 5 min, depending on the power of the computer being used.
- Export matrices of each decay parameter - α2 (%),
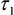 ,
, 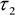 - and the photon counts at each pixel for subsequent analysis in ImageJ.
- and the photon counts at each pixel for subsequent analysis in ImageJ. - Import α2 (%), , and photon count images into ImageJ using the Import Text Image command.
- Use thresholding and the Create Selection command on the photon count image to add a region of interest (ROI) containing the cells to the ROI manager.
- Use the polygon selection tool to create a ROI of the cell nuclei and save to ROI manager. The nuclei typically have altered NAD(P)H fluorescence decay characteristics, due to differences in metabolism to the rest of the cell, so are neglected from the analysis here for simplicity.
- Create a mask from the cell ROI. Select the nuclear ROI when in the mask window and fill with black. Use thresholding to select the resulting image containing white cell bodies and black nuclei. Add this selection to the ROI manager.
- Use the cell body ROI to measure the mean values of ,, , and intensity <I> from the corresponding images.
- Repeat the process for each image, transferring results for each parameter to Excel. For each cell type, calculate the mean (± SE) value of each parameter. Differences in parameter values can be assessed for statistical significance using a Wilcoxon signed-rank test.
- In each cell line, the ratio of NADPH to NADH can be related to the measured parameters using,
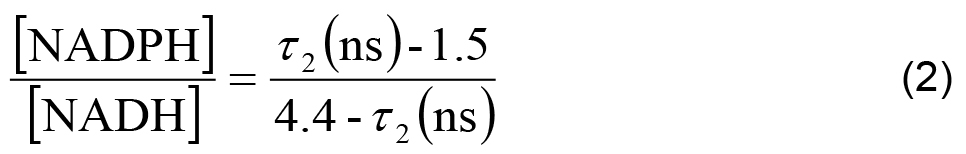
- The measured intensity and the remaining decay parameters can be used to calculate the relative concentrations of enzyme-bound NADH and NADPH using,
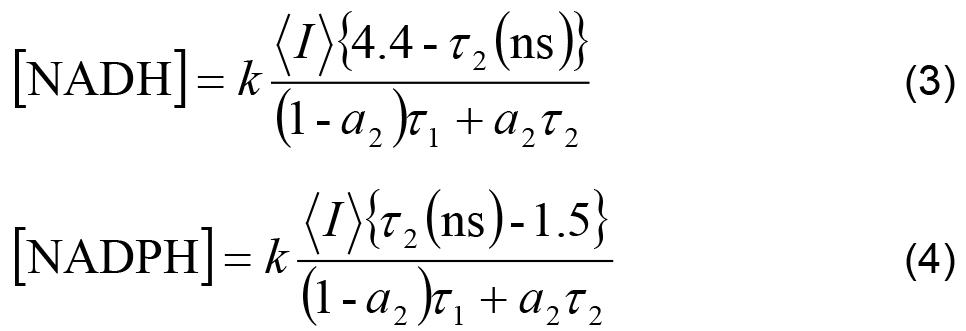
Where,
k is an arbitrary constant shared between all experiments, assuming experimental settings (e.g., laser power) were kept constant.
Notes
- Plating of cells for microscopy
- Coverslips can be sterilised prior to being placed into the 6 well plate. This can be achieved by autoclaving, passing through a flame, or being dipped in ethanol. Sufficient time should be left for the coverslips to cool or for all traces of ethanol to evaporate before the cell suspension is added to the well.
- Different cell lines exhibit different adherence for glass. Cells that attach poorly may require treatment of the coverslips with gelatin, fibronectin or poly-L-lysine. Coverslips can also be purchased pre-treated.
- NAD redox state assay
- The NAD(P)H fluorescence intensity is low relative to extrinsic fluorophores conventionally imaged using confocal microscopy. As such, care should be taken to fully optimise the signal prior to experimental recordings. The on-sample laser power chosen should not be so high as to cause bleaching, and associated photodamage (Tiede and Nichols, 2006), of the detected signal over the ~30 min time period of the experiment. The highest numerical aperture objective available should be chosen, perhaps necessitating the use of an oil immersion lens in an inverted configuration. Laser collimators should be adjusted to maximise contrast and confocal pinholes should be a large as possible, as precise z-resolution is not required in this assay. Finally, the detector gain and offset settings should be chosen in preliminary experiments such that FCCP does not cause the detected signal to reach zero and that rotenone does not cause detector saturation.
- Acquisition settings (e.g., laser power and detector gain) must be kept constant in order to obtain comparable results between technical and biological replicates. Since measured intensities are a linear function of the applied laser power with single-photon excitation, linear correction (normalisation) can be applied if the laser power is modified. Gain on confocal microscopes has a non-linear effect on intensities which makes normalisation impractical if it is adjusted. Ideally, threshold settings during analysis should also be kept constant in a given set of experiments, in particular if absolute levels of NAD(P)H are compared between different conditions or cell lines.
- For assistance in choice of excitation wavelength and filtering, the absorption and emission spectra of NAD(P)H are included in Figure 6, adapted from Patterson et al. (2000).
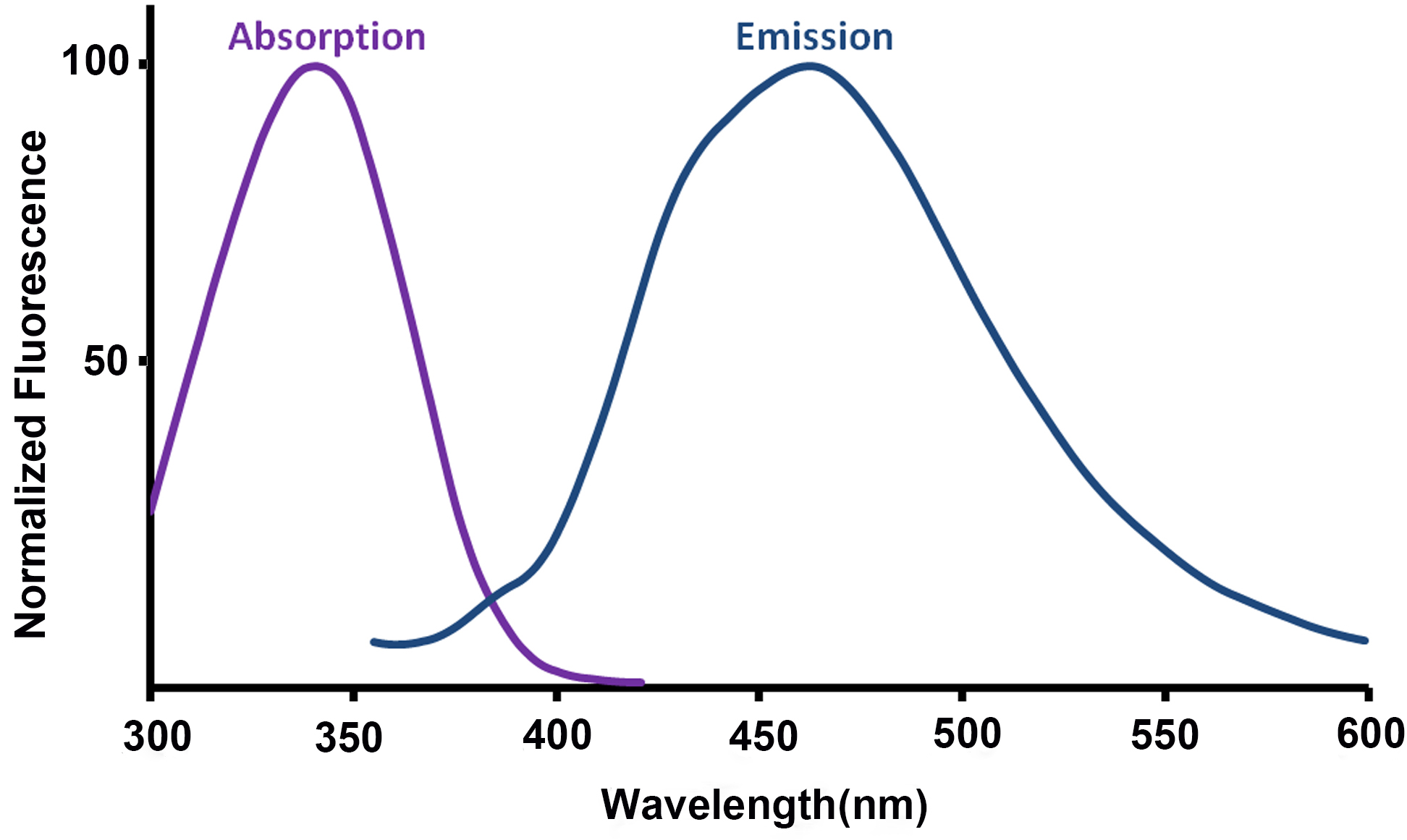
Figure 6. Absorption and emission spectra of NAD(P)H (adapted from Patterson et al., 2000) - The NAD(P)H fluorescence signal should be allowed to reach a steady level following the addition of FCCP or rotenone before the experimental conditions are subsequently altered (addition of the next drug, or termination of the experiment). This may require longer than the 8 min suggested here due to the time taken for the drug to mix around the dish. This process may be aided by pipetting larger volumes of a more dilute solution to reach the final concentration.
- The primary effects of FCCP and rotenone are to oxidise and reduce the NAD pool, respectively. As such, this can be viewed as an assay of the mitochondrial NAD redox state. However, these treatments may have secondary effects on the NADP pool through the action of the mitochondrial nicotinamide nucleotide transhydrogenase (NNT). This will oxidise NADH to reduce NADPH under control and rotenone-treated conditions but may reverse to consume NADPH and replenish the NADH supply with FCCP (Nickel et al., 2015). The qualitative influence on the final result will be minimal as the NAD pool is much larger than the NADP pool, but this caveat should be noted when attempting to draw quantitative conclusions (Blacker and Duchen, 2016).
- Once experimental protocols have been correctly established, this assay is generally highly reproducible, with typical standard deviations of the scaled fluorescence levels across identical experiments of around 2%. This allows around 3-5 repeats to be sufficient to have statistical confidence in the final results.
- NAD(P)H FLIM assay
- As the detectors required for FLIM have single photon sensitivity, their protection from bright light sources is of utmost importance. Shutter assemblies should be purchased with the detectors, blackout curtains over the microscope itself (Figure 1) should be used to protect from room lights and use of mercury lamps in the experiments should be minimised. Seals between the detector and the microscope should be routinely inspected to ensure no light can leak in.
- For the most accurate extraction of fluorescence decay parameters from FLIM data, SPCImage should be supplied with a measurement of the instrument response function of the system. This can be acquired by measuring the fluorescence lifetime profile of a scattering object. For example, we routinely use the second harmonic generation signal of potassium dihydrogen phosphate (KDP) crystals, grown by leaving a molar solution on a coverslip to evaporate overnight.
- This protocol has been written assuming two-photon excitation will be used for FLIM. Pulsed UV lasers exist, allowing NAD(P)H fluorescence lifetime measurements with single photon excitation. However, the pulsed Ti:sapphire laser is ubiquitous in biomedical microscopy facilities in order to perform two-photon intensity imaging on thick samples. As such, FLIM add-ons are typically installed onto these existing systems.
- The models used to interpret NAD(P)H FLIM data in terms of the separate concentrations of NADH and NADPH (equations 2-4) are subject to a number of assumptions (Blacker et al., 2014). The balance of the two cofactors in the enzyme-bound population is assumed to reflect that in the free population, and these species are assumed to possess finite, distinct lifetimes. These models have shown success in unravelling the separate roles of NADH and NADPH in a number of biological contexts (Blacker et al., 2014; Nickel et al., 2015; Tosatto et al., 2016). However, their quantitative accuracy will be increased by ongoing refinements based on enhanced understanding of the contrasting photophysics of the enzyme-bound cofactors, and their relationship to the underlying metabolism.
- Pixel-to-pixel variability exists in the fluorescence lifetimes measured in an NAD(P)H FLIM image due to noise inherent to the TCSPC technique. Extracting the mean lifetime value from a region of interest has been shown to negate the effect of this Poisson noise, reporting the true underlying lifetime value (Blacker et al., 2014). Cell to cell variability in the fluorescence decay parameters also exists, with standard deviations of around 5%. As such, we typically acquire 3-5 images per coverslip over 3-6 coverslips in order to assess statistically significant differences in the fluorescence decay parameters between conditions.
Recipes
- Routine cell culture medium (500 ml)
440 ml Advanced DMEM (500 ml bottle with 60 ml removed)
50 ml fetal bovine serum (10% final concentration)
5 ml GlutaMAXTM (2 mM final concentration)
5 ml penicillin-streptomycin (100 U ml-1 penicillin and 100 mg ml-1 streptomycin final) - Live-cell imaging medium (50 ml)
415 mg DMEM powder
0.5 ml GlutaMAXTM (2 mM final concentration)
225 mg D-(+) glucose (25 mM final concentration)
5.5 mg sodium pyruvate (1 mM final concentration)
119 mg HEPES (10 mM final concentration)
Top up to 50 ml with ultrapure water
Adjust pH to 7.4, increasing using NaOH or decreasing using HCl
Sterilise using syringe filter - ETC perturbations
- FCCP: 2.5 mg in 10 ml ethanol for 1 mM stock solution (stored in freezer). Dilute 25 μl in 5 ml of imaging medium for 5 μM working solution on day of experiment
- Rotenone: 3.9 mg in 10 ml ethanol for 1 mM stock solution (stored in freezer). Dilute 150 μl in 5 ml of imaging medium for 30 μM working solution on day of experiment
Acknowledgments
The use of NAD(P)H autofluorescence for assessing the redox state of live tissues was originated by Britton Chance in the 1950’s (Chance et al., 1962; Chance et al., 1952; Chance and Williams, 1955). These protocols extend this pioneering work onto modern apparatus. We acknowledge support from BBSRC grant BB/L020874/1.
References
- Blacker, T. S. and Duchen, M. R. (2016). Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med 100: 53-65
- Blacker, T. S., Mann, Z. F., Gale, J. E., Ziegler, M., Bain, A. J., Szabadkai, G. and Duchen, M. R. (2014). Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun 5: 3936.
- Chance, B. (1952). Spectra and reaction kinetics of respiratory pigments of homogenized and intact cells. Nature 169(4293): 215-221.
- Chance, B., Cohen, P., Jobsis, F. and Schoener, B. (1962). Intracellular oxidation-reduction states in vivo. Science 137(3529): 499-508.
- Chance, B. and Williams, G. R. (1955). Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem 217(1): 409-427.
- De Ruyck, J., Fameree, M., Wouters, J., Perpete, E. A., Preat, J. and Jacquemin, D. (2007). Towards the understanding of the absorption spectra of NAD(P)H/NAD(P)+ as a common indicator of dehydrogenase enzymatic activity. Chem Phys Lett 450, 119-122.
- Duchen, M. R., Surin, A. and Jacobson, J. (2003). Imaging mitochondrial function in intact cells. Methods Enzymol 361: 353-389.
- Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1-13.
- Nickel, A. G., von Hardenberg, A., Hohl, M., Loffler, J. R., Kohlhaas, M., Becker, J., Reil, J. C., Kazakov, A., Bonnekoh, J., Stadelmaier, M., Puhl, S. L., Wagner, M., Bogeski, I., Cortassa, S., Kappl, R., Pasieka, B., Lafontaine, M., Lancaster, C. R., Blacker, T. S., Hall, A. R., Duchen, M. R., Kastner, L., Lipp, P., Zeller, T., Muller, C., Knopp, A., Laufs, U., Bohm, M., Hoth, M. and Maack, C. (2015). Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab 22(3): 472-484.
- Osellame, L. D., Blacker, T. S. and Duchen, M. R. (2012). Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab 26(6): 711-723.
- Patterson, G. H., Knobel, S. M., Arkhammar, P., Thastrup, O. and Piston, D. W. (2000). Separation of the glucose-stimulated cytoplasmic and mitochondrial NAD(P)H responses in pancreatic islet β cells. Proc Natl Acad Sci U S A 97(10): 5203-5207.
- Pawley, J. (2012). Handbook of Biological Confocal Microscopy. Springer.
- Pollak, N., Dolle, C. and Ziegler, M. (2007). The power to reduce: pyridine nucleotides--small molecules with a multitude of functions. Biochem J 402(2): 205-218.
- Tiede, L. M. and Nichols, M. G. (2006). Photobleaching of reduced nicotinamide adenine dinucleotide and the development of highly fluorescent lesions in rat basophilic leukemia cells during multiphoton microscopy. Photochem Photobiol 82(3): 656-664.
- Tosatto, A., Sommaggio, R., Kummerow, C., Bentham, R. B., Blacker, T. S., Berecz, T., Duchen, M. R., Rosato, A., Bogeski, I., Szabadkai, G., Rizzuto, R. and Mammucari, C. (2016). The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol Med 8(5): 569-585.
- Ying, W. (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10(2): 179-206.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Blacker, T. S., Berecz, T., Duchen, M. R. and Szabadkai, G. (2017). Assessment of Cellular Redox State Using NAD(P)H Fluorescence Intensity and Lifetime. Bio-protocol 7(2): e2105. DOI: 10.21769/BioProtoc.2105.
Category
Cancer Biology > Invasion & metastasis > Cancer therapy
Cancer Biology > Cellular energetics > Cell biology assays > Metabolism
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.