- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Golden Gate-based Protocol for Assembly of Multiplexed gRNA Expression Arrays for CRISPR/Cas9


(*contributed equally to this work) Published: Vol 6, Iss 23, Dec 5, 2016 DOI: 10.21769/BioProtoc.2059 Views: 27457
Reviewed by: Renate WeizbauerGal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The CRISPR (clustered regularly interspaced short palindromic repeats)-associated protein 9 (Cas9) has become the most broadly used and powerful tool for genome editing. Many applications of CRISPR-Cas9 require the delivery of multiple small guide RNAs (gRNAs) into the same cell in order to achieve multiplexed gene editing or regulation. Using traditional co-transfection of single gRNA expression vectors, the likelihood of delivering several gRNAs into the same cell decreases in accordance with the number of gRNAs. Thus, we have developed a method to efficiently assemble gRNA expression cassettes (2-30 gRNAs) into one single vector using a Golden-Gate assembly method (Vad-Nielsen et al., 2016). In this protocol, we describe the detailed step-by-step instructions for assembly of the multiplexed gRNA expression array. The gRNA scaffold used in our expression array is the gRNA 1.0 system for the Cas9 protein from Streptococcus pyogenes driven by the human U6 promoter.
Background
The broadened CRISPR toolbox based on wild-type Cas9 or nuclease-deficient Cas9 (dCas9) has greatly facilitated genome/epigenome editing and regulation in all organisms. Multiplexed gene editing or regulation requires simultaneous expression of several gRNAs in the same cell. The traditional way of delivering several gRNAs into cells is based on either co-transfection of individual gRNA expression vectors or generation of a vector carrying multiple gRNA expression cassettes using traditional cloning; a process which is extremely time consuming. Another way of generating a vector containing multiple gRNA expression cassettes is based on gene synthesis, which is costly and only applicable when working with a very limited number of gRNA expression cassettes. The current protocol is based on Golden Gate cloning which can be used to assemble up to 30 individual gRNA expression cassettes into a single vector within 7 days (Figure 1), with each cassette being driven by an individual human U6 promoter. In our study, we have validated the applicability of this system in both human and porcine cells, but it is in principle compatible with applications in any other organisms that can utilize the human U6 promoter. Compared with existing methods, our method is cost effective, rapid (7 days) and flexible (applicable with any gRNAs that do not contain a BbsI, BsaI or BsmBI recognition site). Many applications of CRISPR/Cas9 may benefit from using our system, including multiplexed gene knockout by CRISPR/SpCas9, multiplexed gene inhibition by CRISPRi, and multiplexed gene activation by CRISPRa.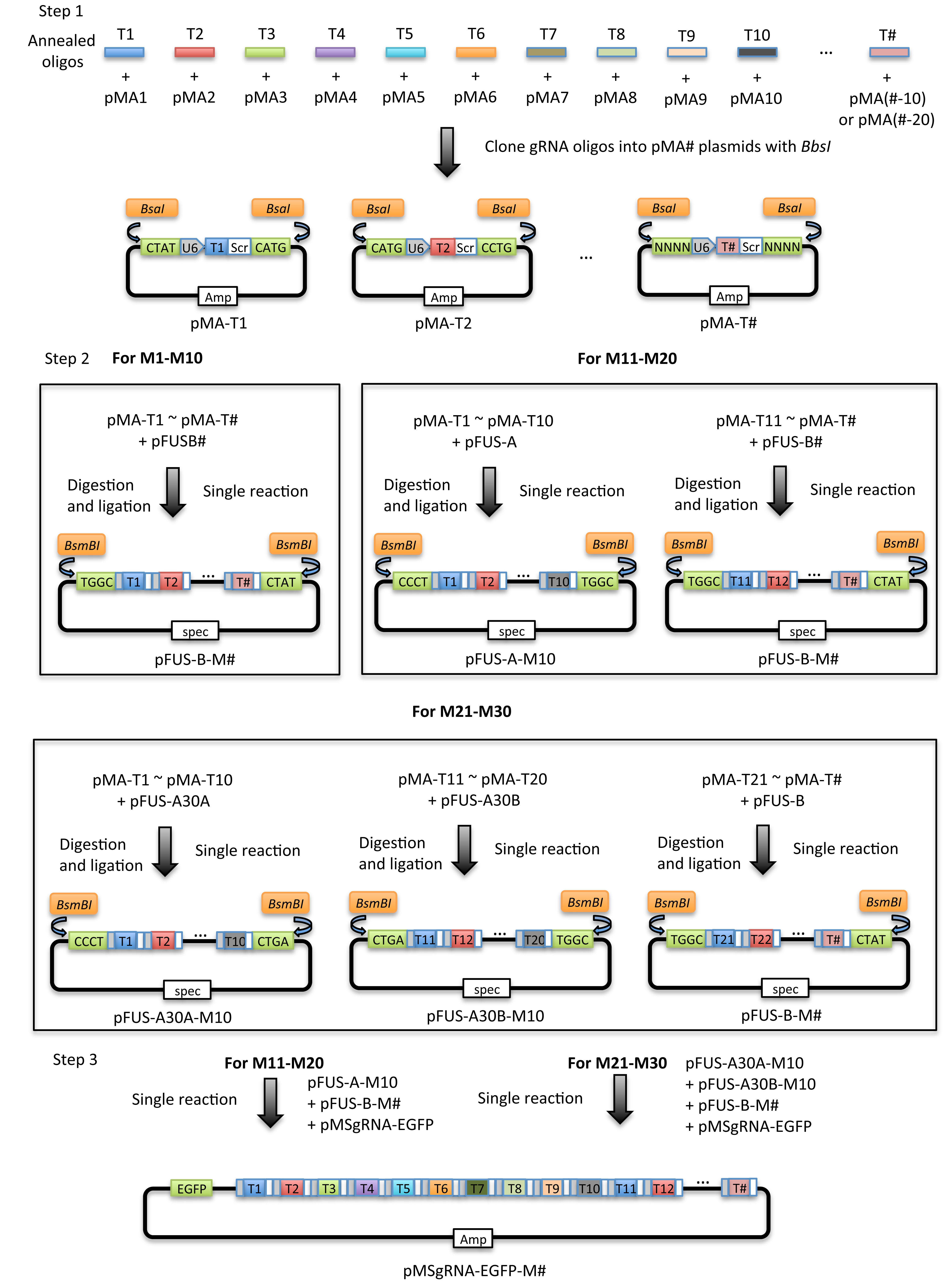
Figure 1. Schematic illustration of the principle of the current protocol. The current protocol is carried out in 2-3 major steps which vary depending on the number of gRNA expression cassettes to be assembled. Step 1: The gRNA oligonucleotides (T#) are cloned into individual modular single gRNA expression vectors (pMA-SpCas9-g#). Step 2: The individual gRNA expression vectors (pMA-T#) are assembled into 1-3 array vectors depending on the total number of gRNAs. Step 3: For assembly of 11-30 gRNA expression cassettes, 2 to 3 individual array vectors are subjected to a second round of assembly to yield the final EGFP expressing vector (pMsgRNA-EGFP).
Materials and Reagents
- 200 µl PCR tubes
- 1.5 ml Eppendorf tubes
- 10 or 100 µl pipette tips
- Competent E. coli cells (No particular preference, but should be recombination deficient)
- The modular gRNA plasmids (available from Addgene, see Table 1 for corresponding Addgene plasmid IDs)
- The pFUS-B1 to pFUS-B10, pFUS-A, pFUS-A30A and pFUS-A30B plasmids (available from Addgene, Golden Gate TALEN and TAL Effector Kit 2.0) (Addgene, catalog number: 1000000024 )
- NEB buffer 2 (New England BioLabs, catalog number: B7002S )
- BbsI (FastDigest) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD1014 )
- T4 DNA ligase (5 U/μl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EL0014 )
- Distilled H2O
- Ampicillin (Sigma-Aldrich, catalog number: A1593 )
- LB medium
- dNTP
- DreamTaq or other equivalent DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K1072 )
- Agarose
- Plasmid prep mini kit (No particular preference, we used the Nucleo Spin plasmid easy pure kit from MACHEREY-NAGEL, catalog number: 740727 )
- BsaI (BpiI) (FastDigest) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0293 )
- Plasmid-SafeTM ATP-Dependent DNase (Epicentre, catalog number: E3101K , ATP is included in this kit)
- Spectinomycin (Sigma-Aldrich, catalog number: PHR1441 )
- X-gal (Sigma-Aldrich, catalog number: B4252 )
- IPTG (dissolved IPTG in water) (Sigma-Aldrich, catalog number: I6758 )
- AflII (BspTI) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0834 )
- XbaI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0684 )
- BsmBI (Esp3I) (FastDigest) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0454 )
- Universal primers for PCR screening (see Table 1)
Table 1. Plasmids and primers. Plasmids developed for this protocol are available from Addgene with corresponding Addgene IDs. Single modular plasmids (pMA-SpCas9-g1 to pMA-SpCas9-10) for cloning gRNA oligonucleotides into individual gRNA expression plasmids. Array plasmid (pMA-MsgRNA-EGFP) for assembly of gRNA expression array containing 11-30 gRNA expression cassettes. Primer sequences used in this protocol for screening of assembled array plasmids.
Note: The Universal U6 and Scr primers have been modified to suit the PCR conditions in this protocol.Plasmid Addgene ID pMA-MsgRNA-EGFP 80794 pMA-SpCas9-g10 80793 pMA-SpCas9-g9 80792 pMA-SpCas9-g8 80791 pMA-SpCas9-g7 80790 pMA-SpCas9-g6 80789 pMA-SpCas9-g5 80788 pMA-SpCas9-g4 80787 pMA-SpCas9-g3 80786 pMA-SpCas9-g2 80785 pMA-SpCas9-g1 80784 Primers Sequences (5’-) Universal U6 Forward ATAAGGATCCGGTCTCGCTATGAGGGCCTATTTCCCATG Universal Scr Reverse ATAATGTACAGGTCTCCCATGTAACTTGCTATTTCTAGCTC
Equipment
- Heating block (Grant Instruments, model: QBD2 )
- Microcentrifuge (Eppendorf, model: Centrifuge 5424 )
- 37 °C Thermo incubator (Labnet International, model: 211DS )
- 37 °C shaking incubator (environmental incubator shaker G24 ) (Eppendorf, New Brunswick Scientific, model: G24)
- Thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, model: Veriti® 96 well thermal cycler )
- DNA electrophoresis apparatus (Bio-Rad Laboratories, model: Wide mini-sub cell GT )
- Nanodrop (Thermo Fisher Scientific, model: Nanodrop 1000 Spectrophotometer )
Procedure
- Preparation: design and order CRISPR gRNA oligos
- Design target gRNA oligonucleotides using any of the online designing tools such as the optimized CRISPR design tool (http://crispr.mit.edu/).
Notes: - Because the gRNA expression cassette utilizes a human U6 promoter, the first nucleotide of the CRISPR target site should preferably be a ‘G’ to ensure efficient transcription.
- It is also very important to exclude gRNA oligonucleotides that contain any of the following type IIS restriction sites: BbsI, BsaI, and BsmBI.
- For target sites starting with a ‘G’, add the following four nucleotide overhangs to the sense (SS) and antisense (AS) gRNA oligonucleotides:
SS: 5’CACC(N20)
AS: 5’AAAC(N20) - For target sites not stating with a ‘G’, add the following overhangs:
SS: 5’CACCG(N20)
AS: 5’AAAC(N20)C - Order oligonucleotides from any qualified company, dilute the oligonucleotides to a stock concentration of 100 pmol/µl (µM) and store at -20 °C. (Standard desalted purification of the oligos is sufficient).
- Design target gRNA oligonucleotides using any of the online designing tools such as the optimized CRISPR design tool (http://crispr.mit.edu/).
- Annealing of gRNA oligos
- Anneal the sense and antisense gRNA oligonucleotides for each gRNA in separate 1.5 ml EP tubes:
Sense gRNA oligo: 1 µl (100 pmol/µl)
Antisense gRNA oligo: 1 µl (100 pmol/µl)
10x NEB buffer 2: 2 µl
Add ddH2O to a final volume of 20 µl. - Denature the mixture at 95 °C for 5 min in a heating block.
- Place the heating block on the lab bench and slowly anneal oligonucleotides until mixture is at room temperature (which takes approximately 1-2 h).
- Quick-spin the annealed gRNA oligonucleotides to the bottom of the tubes and store at -20 °C for later use or proceed with the following experimental procedure.
- Anneal the sense and antisense gRNA oligonucleotides for each gRNA in separate 1.5 ml EP tubes:
- Vector cloning
Using this protocol, the generation of the gRNA expression array vector is accomplished in 5 days (for 1 to 10 gRNA expression cassettes) to 7 days (for 11 to 30 cassettes). See schematic in Figure 2 for an overview. In the protocol, the location of a target site in the CRISPR expression array is referred to as T# (#, an integer between 1 and 30 denoting the order of the gRNAs). The protocol utilizes 10 individual gRNA expression cassettes that are tandemly assembled into a set of ‘array’ vectors depending on the number of cassettes, where each array vector contains up to 10 cassettes. When assembling 11-30 cassettes, multiple array vectors are finally assembled into a single vector. The selection of plasmids for assembly is described in step C3c, Table 3. A schematic illustration is given in Figure 1.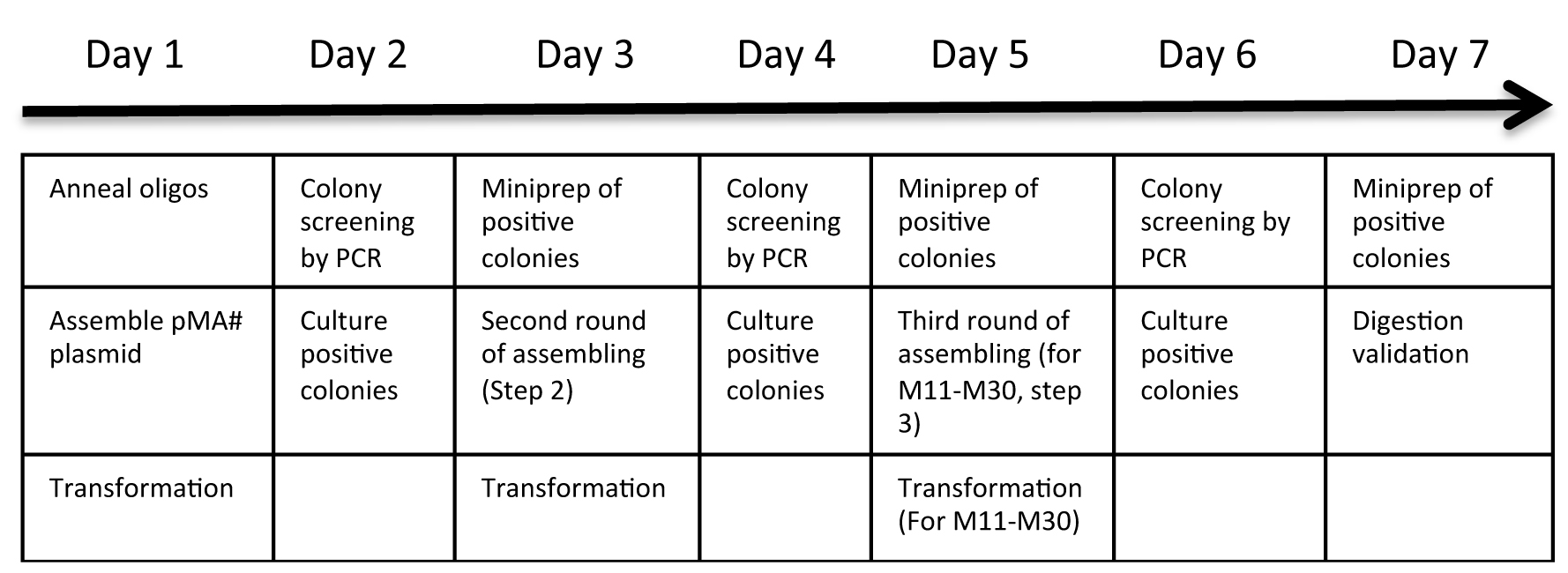
Figure 2. Protocol overview. Day to day overview of the protocol. For assembly 1 to 10 gRNA expression cassettes, the protocol is completed after miniprep of positive colonies at day 5. For assembling 11 to 30 gRNA expression cassettes, an additional round of assembly is performed at day 5 and completed after day 7.- Day 1. Ligation of gRNA oligonucleotides into individual modular single gRNA expression vectors
- Select pMA plasmids for annealed gRNA oligonucleotides T1-TN (N ≤ 30) generated in step B4 according to the table below (pMA-SpCas9-g1 for T1, pMA-SpCas9-g2 for T2, …, pMA-SpCas9-g1 for T11, pMA-SpCas9-g2 for T12, …, pMA-SpCas9-g1 for 21, pMA-SpCas9-g2 for T22, etc.) (Table 2).
Table 2. Assembly scheme of modular single gRNA expression vectors and annealed gRNA oligonucleotides. This table illustrates how to choose the right single modular plasmids (pMA-SpCas9-g1 to pMA-SpCas9-10) for cloning of the gRNA oligonucleotides into individual gRNA expression plasmids (named pMA-T# after successful cloning). The number of each gRNA represents the position of the gRNA in the tandem gRNA expression array.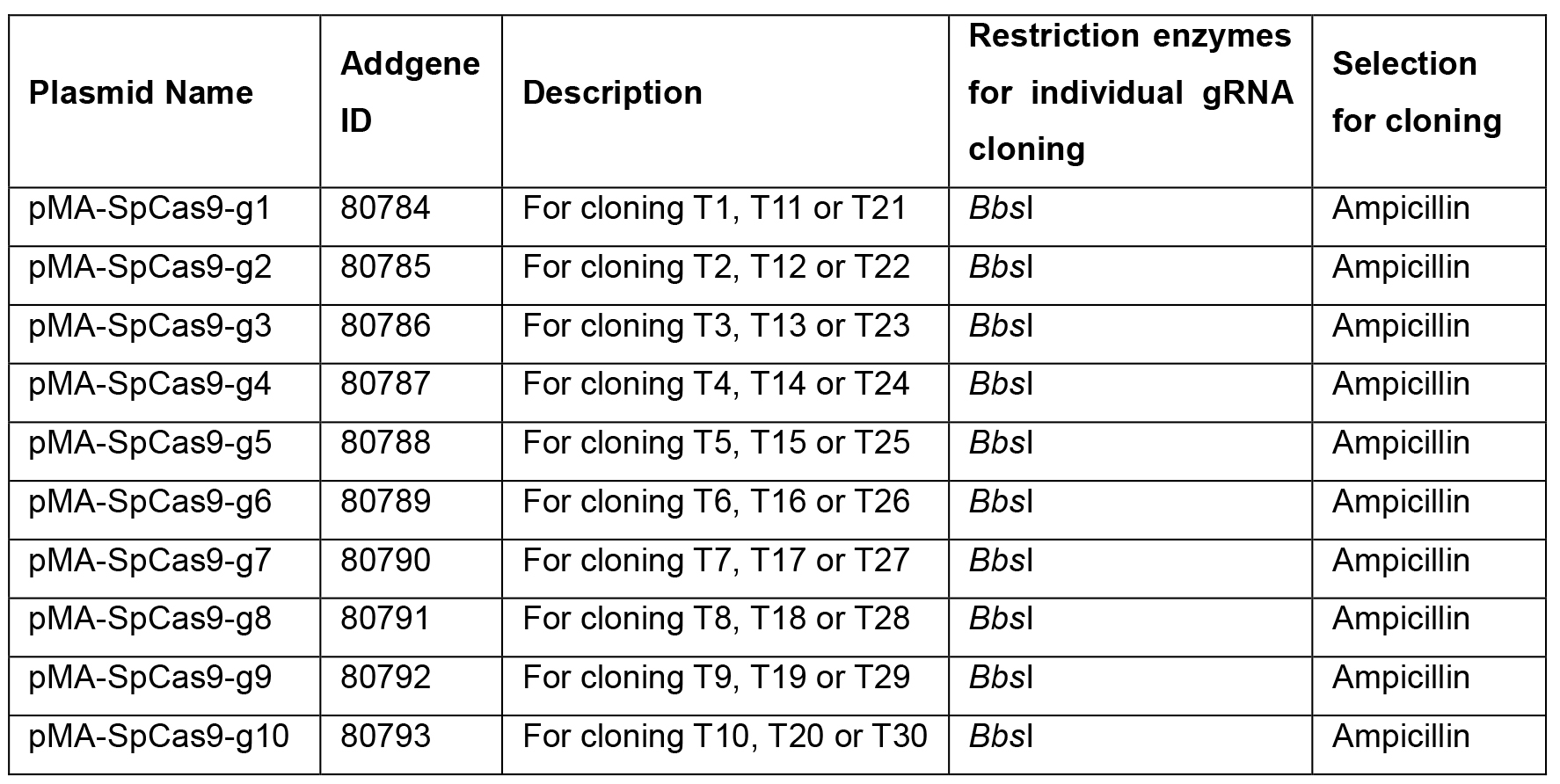
- Prepare assembly reaction in individual PCR tubes containing:
- 100 ng modular single gRNA expression plasmid (pMA-SpCas9-g#).
- 1 µl of annealed gRNA oligonucleotide from step B4 (T#).
- 1 µl BbsI restriction enzyme.
- 1 µl T4 DNA ligase.
- 2 µl 10x T4 ligase buffer (to a final concentration of 1x).
- Nuclease-free water up to 20 µl total reaction volume.
- Incubate the reactions in a thermal cycler with the following conditions:
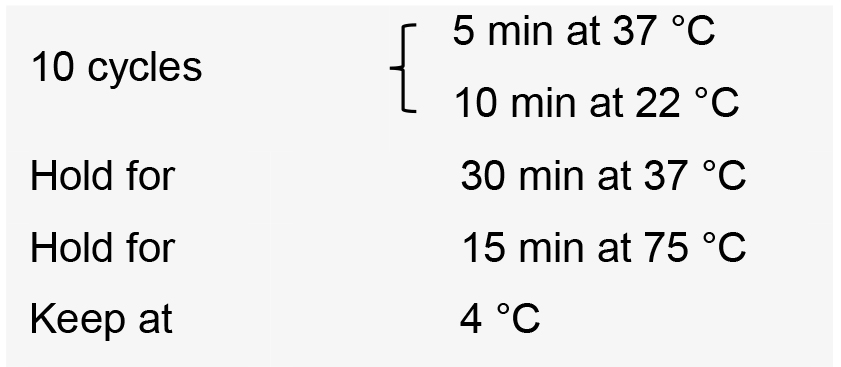
- Transform competent E. coli using 2 µl of the ligation product.
Note: Protocols for generation of competent E. coli and transformation are available from the authors upon request. - Plate (1/10) transformed cells on a 10 cm LB agar plate containing 50 µg/ml ampicillin.
Note: Do not over-plate the cells. - Incubate agar plates at 37 °C overnight.
- Day 2. 1st colony screening and culture
- Pick two to three colonies from each transformation using a 10 or 100 µl pipette tip.
- Dip the pipette tip in 100 µl LB medium (50 µg/ml ampicillin) to start initial culture. Transfer the pipette tip to a PCR tube containing 30 µl nuclease-free water and pipette up and down to release cells.
Note: The initial culture and PCR tube should be labeled with the same ID. If working with over 10 gRNAs, we normally use two sets of 200 µl PCR tubes, one set for the initial culture and the other set for the cell lysate. - Place the initial culture tubes in a 37 °C incubator.
- Place the PCR tubes in a thermal cycler at 98 °C for 10 min to lyse cells.
- Perform gRNA PCR screenings of cell lysates. Add to each reaction:
- 1 µl transformed E. coli lysate.
- 5 pmol universal U6 Forward primer (Table 1).
- 5 pmol antisense oligonucleotide of the gRNA in question.
- 0.4 µl dNTP (10 mM).
- 0.1 µl DreamTaq polymerase (or similar).
- 2 µl 10x DreamTaq buffer (to final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
- 1 µl transformed E. coli lysate.
- Incubate the PCR reactions in a thermal cycler with the following conditions:
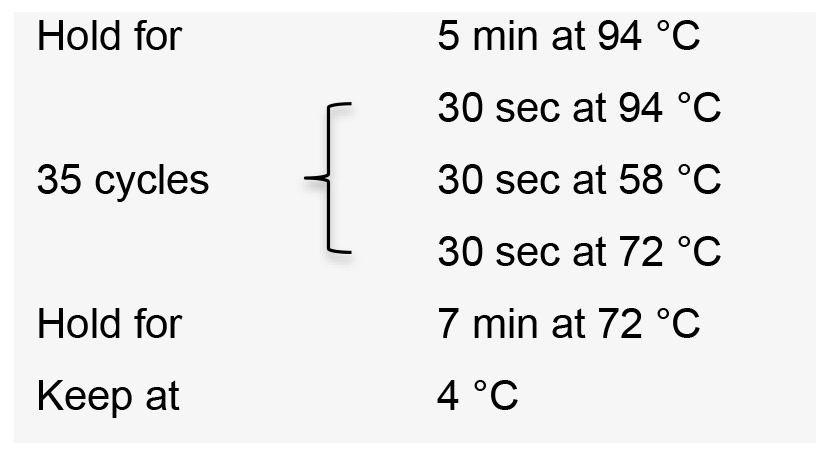
- Visualize PCR product on a 1% agarose gel by electrophoresis. The expected amplicon size is approximately 270 bp.
- Select one positive colony from each transformation and transfer 50 µl of initial culture to individual tubes containing 5 ml LB medium with 50 µg/ml ampicillin.
- Incubate overnight at 37 °C in an orbital shaker.
- Pick two to three colonies from each transformation using a 10 or 100 µl pipette tip.
- Day 3. Plasmid prep and assembly of individual gRNA expression cassettes into the ‘array’ plasmids
- Isolate the plasmid DNA (named pMA-T#) from the overnight cultures using a commercial miniprep kit according to the manufacturer’s instructions.
- Validate the single gRNA expression plasmids by Sanger sequencing using the universal U6 primer (recommended).
- Select array plasmids for 1st assembly of single gRNA expression plasmids according to Table 3:
Table 3. Selection of array plasmids for assembly of pMA-T# vectors. Selection of array plasmids depends on the number of gRNA expression cassettes to be assembled. For assembly of 1-10 gRNA cassettes (Left), one of the pFUS-B# array plasmids is selected depending on the number of gRNA cassettes. When assembling 11-20 gRNA cassettes (Middle), pFUS-A is selected for the assembly of gRNA cassettes 1 to 10. For the remaining cassettes (≤ 10), one of the pFUS-B# array plasmids is selected depending on the number of gRNA cassettes. For assembly of 21-30 gRNA cassettes (Right), pFUS-A30A is used for assembly of the first 10 gRNA expression cassettes. pFUS-A30B is used for assembling gRNA cassettes 11 to 20. The remaining cassettes (≤ 10) are cloned into a pFUS-B# array plasmid depending on the number of gRNA cassettes.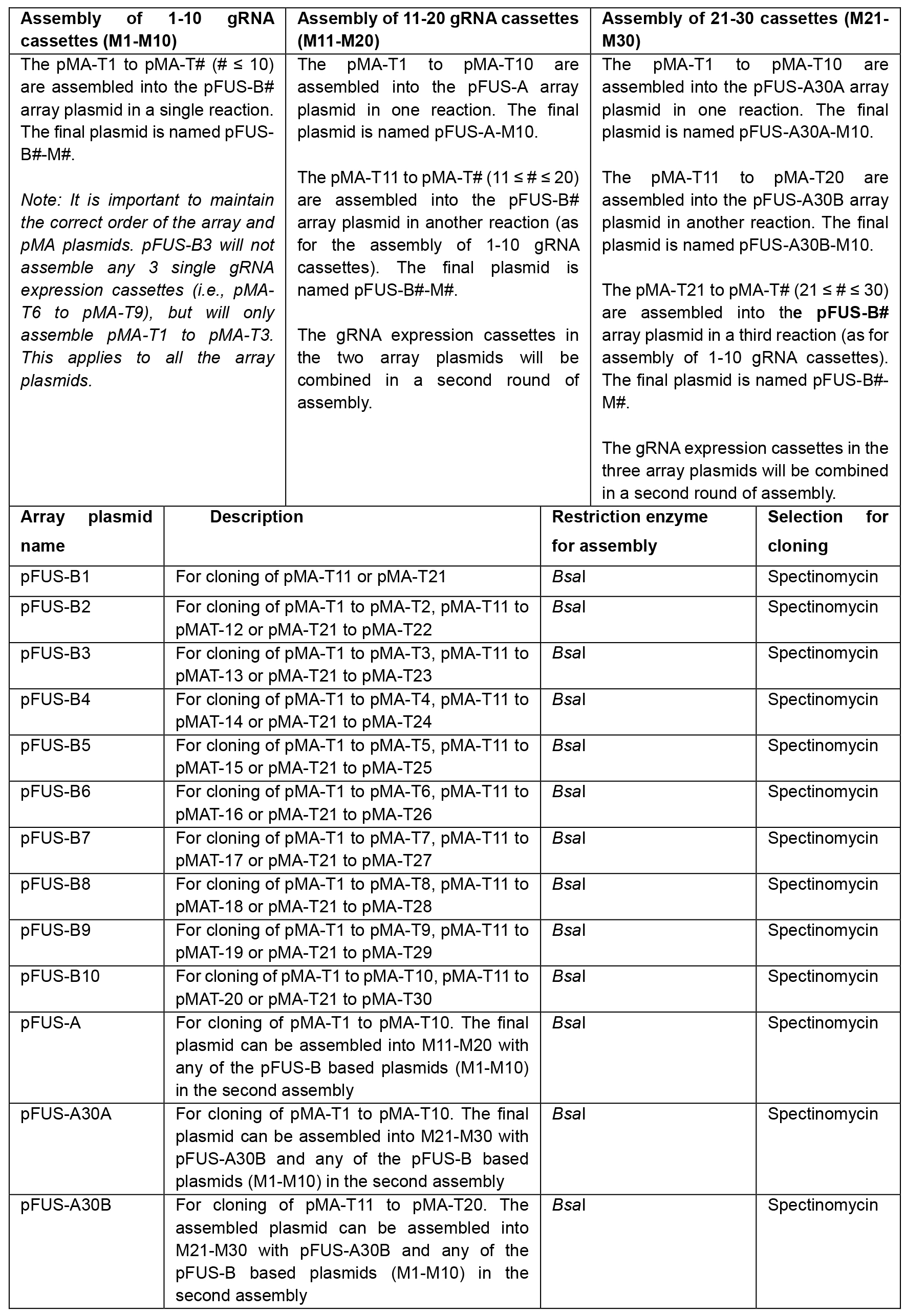
- Set up Golden Gate assembly reactions in individual PCR tubes containing:
- 30 ng array plasmid.
Note: The plasmid amount has been optimized. - 50 ng of each individual modular single gRNA plasmid (maximum of 10 plasmids).
- 1 µl BsaI restriction enzyme.
- 1 µl T4 DNA ligase.
- 2 µl 10x T4 ligase buffer (to a final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
- 30 ng array plasmid.
- Incubate the reactions in a thermal cycler with the following conditions:
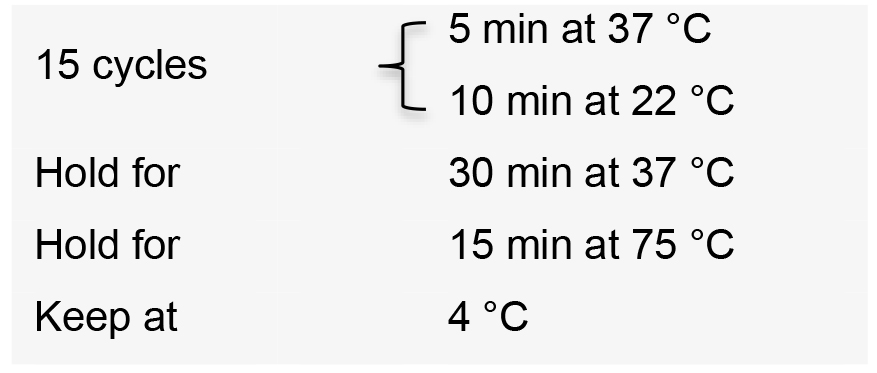
- Add 1 µl 25 mM ATP and 1 µl plasmid safe DNase to the reaction.
Note: Plasmid safe DNase treatment digests all unligated linear dsDNA fragments, incomplete assembly products and linearized vectors. This step minimizes the risk of in vivo recombination of linearized dsDNA fragments in the competent cell. This step is very important when assembling more than five gRNA expression cassettes. - Incubate the reactions in a thermal cycler for 1 h at 37 °C.
- Transform competent E. coli cells using 2 µl of the assembled product.
- Plate (1/20) of the transformed cells on 10 cm LB agar plates containing 50 µg/ml spectinomycin as well as 8 µl 100 mg/µl X-gal and 8 µl 0.5 M IPTG for blue/white colony screening.
- Incubate agar plates at 37 °C overnight.
- Isolate the plasmid DNA (named pMA-T#) from the overnight cultures using a commercial miniprep kit according to the manufacturer’s instructions.
- Day 4. 2nd colony screening and culture
- Inspect plates for positive white colonies. The expected ratio of white to blue colonies is at least 25% for successful assembly of 10 gRNA expression cassettes and will increase gradually with a decreased number of gRNA expression cassettes (Figure 3).
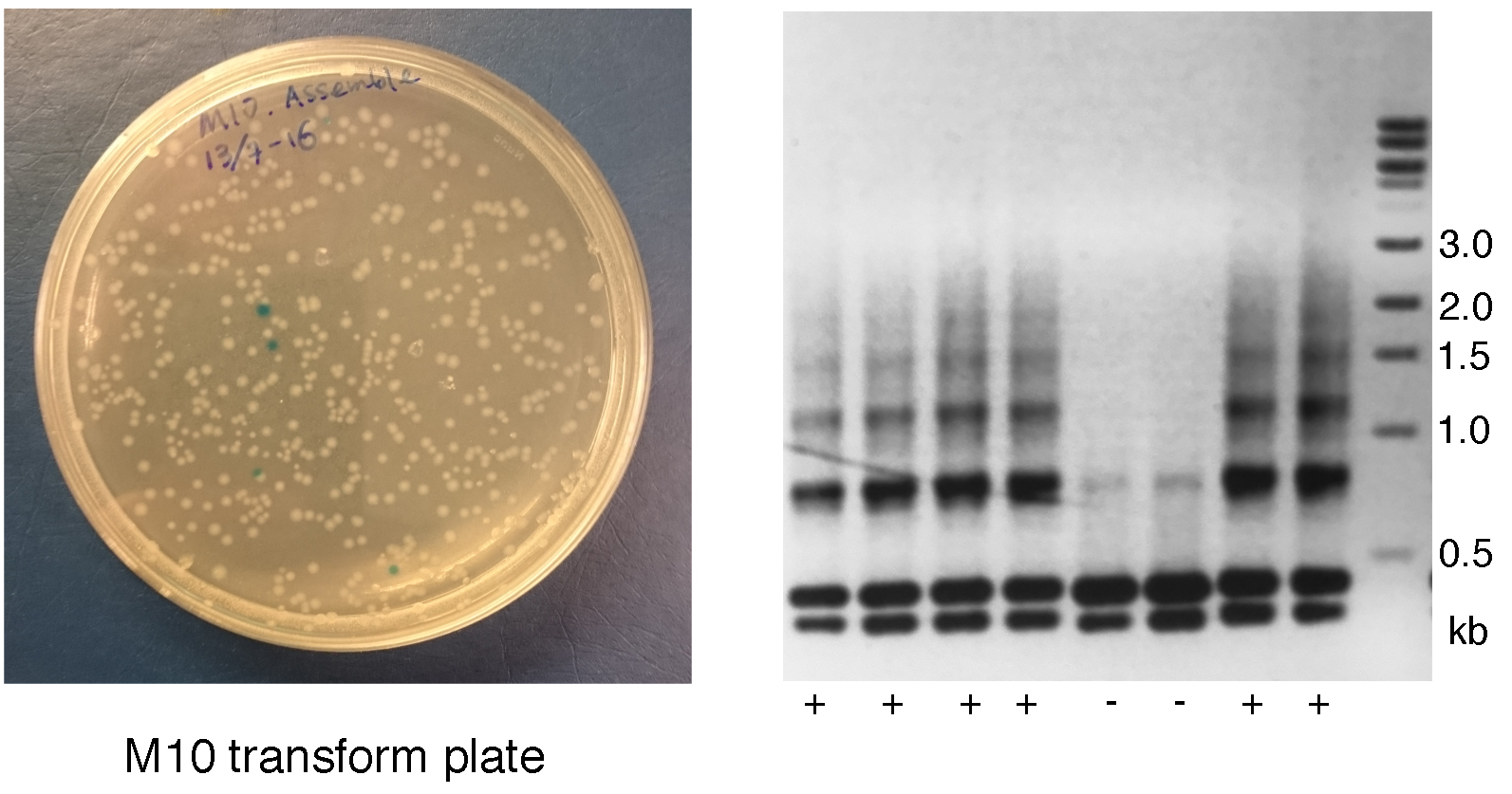
Figure 3. Inspection and screening of first round of assembling. Left: The transformation plate for M10. White colonies contain vectors with insertions. Blue colonies contain empty vectors. Right: Universal PCR for M10 screening of 8 white colonies. Lanes 1-4 and 7-8 show multiple bands resembling a ladder indicative of a positive colony. Lanes 5 and 6 only show the two dominant bands without a ladder indicative of an incorrect insertion. - Pick two to three white colonies from each transformation as described for Day 2.
- Set up PCR screening reactions for all picked colonies in individual PCR tubes containing:
- 1 µl transformed E. coli lysate.
- 5 pmol universal U6 Forward primer (Table 1).
- 5 pmol universal Scr Reverse primer (Table 1).
- 0.4 µl dNTP.
- 0.1 µl DreamTaq polymerase (or similar).
- 2 µl 10x DreamTaq buffer (to a final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
- Incubate the PCR reactions in a thermal cycler with the following conditions:
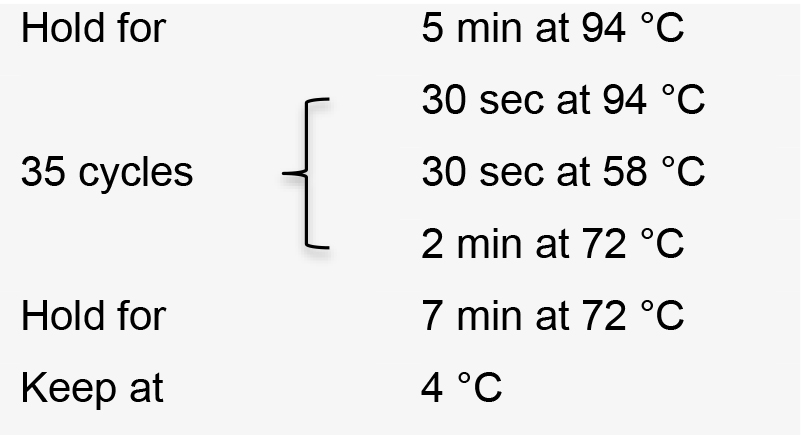
- Visualize PCR product on a 1% agarose gel by electrophoresis. The correct amplification pattern should be a ‘ladder’ with a dominant band at approximately 400 bp and an increase of 392 bp for each ‘step’ corresponding to each additional gRNA expression cassette included in the amplification (Figure 3B).
- Select one positive colony from each transformation and transfer 50 µl of initial culture to individual tubes containing 5 ml LB medium with 50 µg/ml spectinomycin.
- Incubate overnight at 37 °C in an orbital shaker.
- Day 5. Plasmid DNA prep and 2nd assembly for plasmids containing 11 to 30 expression cassettes
- Isolate the plasmid DNA from the overnight cultures using a commercial miniprep kit according to the manufacturer’s instructions.
- Validate the assembled array plasmids by restriction enzyme digestion. In a 1.5 ml EP tube, add:
- 0.5 µg assembled array plasmid DNA.
- 1 µl AflII restriction enzyme.
- 1 µl XbaI restriction enzyme.
- 2 µl 10x reaction buffer (to a final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
- 0.5 µg assembled array plasmid DNA.
- Visualize digestion product on a 1% agarose gel by electrophoresis.
Note: The expected size of the released expression cassette is approximately 4 kb for 10 cassettes (~400 bp per gRNA expression cassette). We strongly recommend generating a theoretical sequence map for each multiplex vector. The RFLP results will be different if there are AflII or Xbal recognition sites in any of the gRNA guide sequences. - For further validation of the assembled array plasmid, set up guide specific PCRs for all gRNA expression cassettes present in the array vector in individual PCR tubes.
Add to each PCR reaction:- 1 ng array plasmid DNA.
- 5 pmol sense oligonucleotide of the specific gRNA in pMA-TN as the forward primer.
- 5 pmol antisense oligonucleotide of the next specific gRNA in pMA-TN+1 as the reverse primer.
- 0.4 µl dNTP.
- 0.1 µl DreamTaq polymerase (or similar).
- 2 µl 10x DreamTaq buffer (to a final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
- 1 ng array plasmid DNA.
- Incubate the PCR reactions in a thermal cycler with the following conditions:
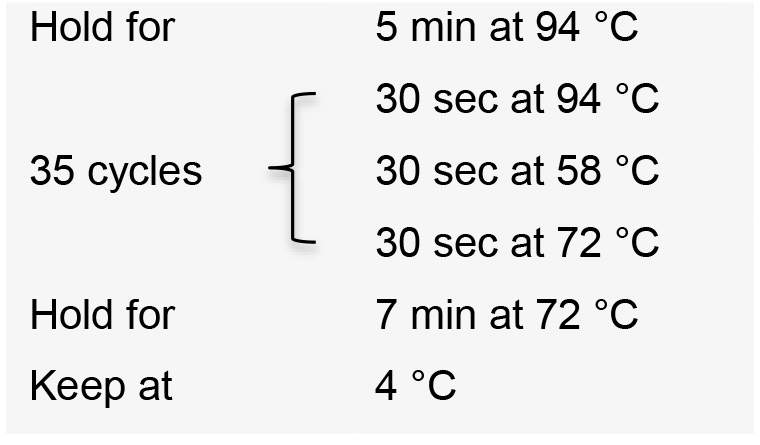
- Visualize PCR product on a 1% agarose gel by electrophoresis. The expected amplicon sizes are approximately 400 bp.
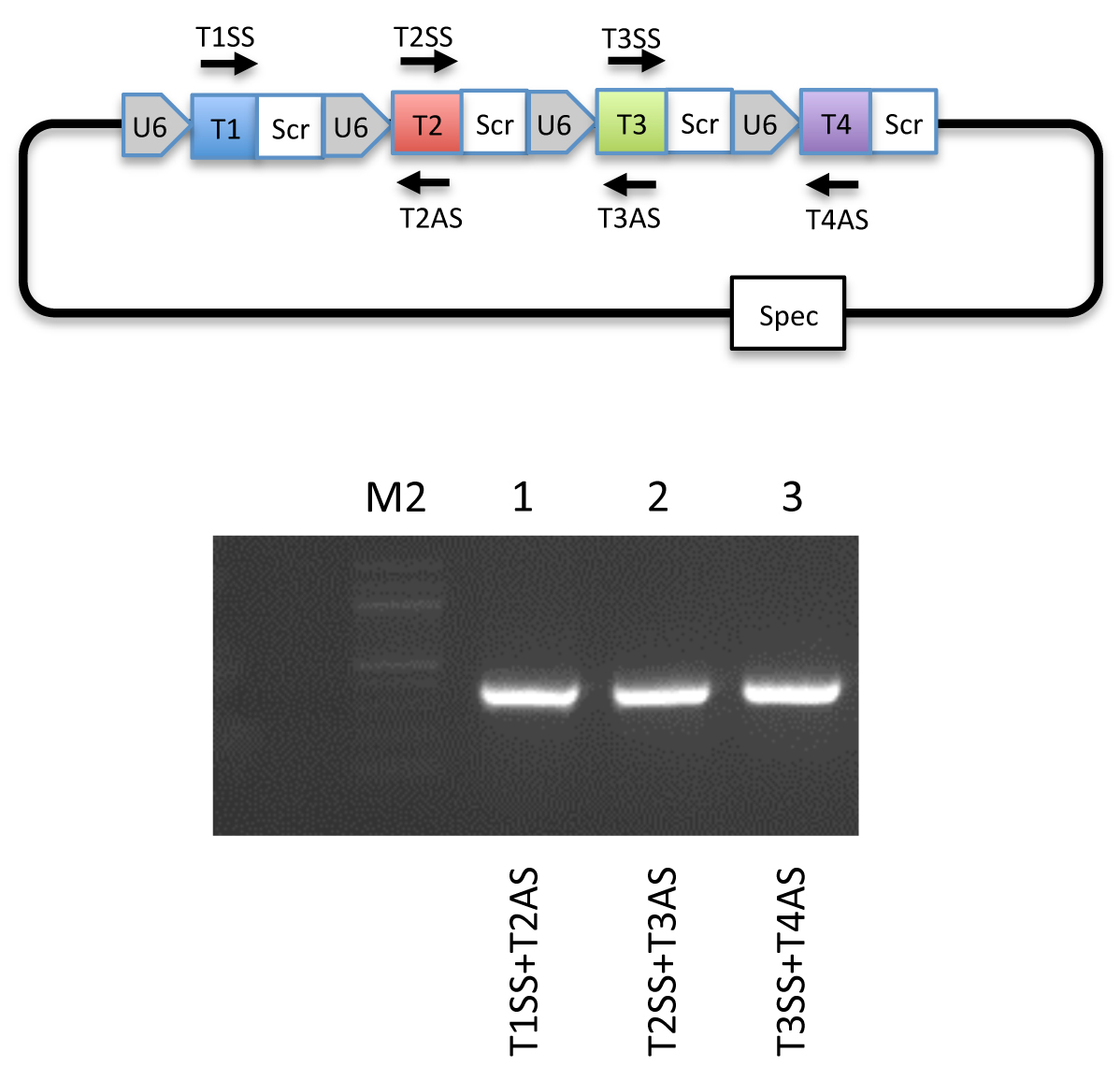
Figure 4. Guide specific PCR validation. Top: Schematic illustration of primers used for guide specific PCR. Bottom: An example of a guide specific PCR using a pFUS-B4-M4 plasmid visualized on a 1% agarose gel. - Set up 2nd assembly of array plasmids in a PCR tube (only for assembly of 11-30 cassettes) containing:
- 50 ng DNA of each assembled array vector (pFUS-A-M10 + pFUS-B#-M# OR pFUS-A30A-M10 + pFUS-A30B-M10 + pFUS-B#-M#).
- 50 ng DNA of array plasmid pMsgRNA-EGFP (Table 1).
- 1 µl BsmBI restriction enzyme.
- 1 µl T4 DNA ligase.
- 2 µl 10x T4 ligase buffer (to a final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
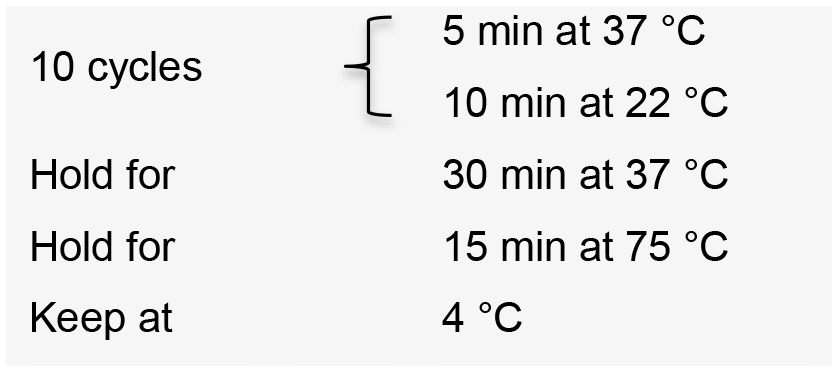
- Incubate the reaction in a thermal cycler with the following conditions:
- Transform competent E. coli cells using 2 µl of the ligation product.
- Plate (1/10) of the transformed cells on a 10 cm LB agar plate containing 50 µg/ml ampicillin as well as 40 μl of 20 mg/ml X-gal and 40 μl of 0.1 M IPTG for blue/white colony screening.
- Incubate agar plate at 37 °C overnight.
- Day 6. 3rd colony screening and culture
- Inspect plate for positive white colonies. The expected ratio of white to blue colonies is > 80% for successful assembly.
- Pick two to three white colonies as described for Day 2.
- Set up overlap PCR screening reactions for each assembled section of gRNA expression cassettes for all picked colonies. To individual PCR tubes, add:
- 1 µl transformed E. coli lysate.
- 5 pmol sense oligonucleotide of the last gRNA (T10 or T20) in the first or second expression cassette array as the forward primer.
- 5 pmol antisense oligonucleotide of the first gRNA (T11 or T21) in the second or third expression cassette array as the reverse primer.
- 0.4 µl dNTP.
- 0.1 µl DreamTaq polymerase (or similar).
- 2 µl 10x DreamTaq buffer (to a final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
- 1 µl transformed E. coli lysate.
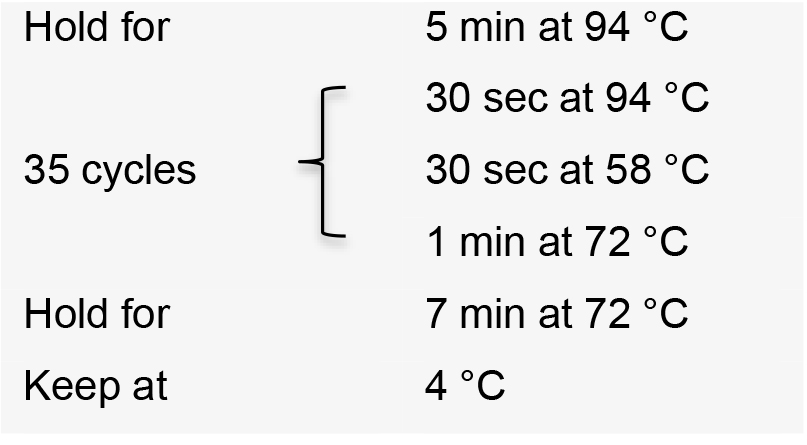
- Incubate the PCR reactions in a thermal cycler with the following conditions:
- Visualize PCR product on a 1% agarose gel by electrophoresis. The expected amplicon size is approximately 460 bp.
- (Optional) Perform overlap PCRs for all neighboring gRNAs.
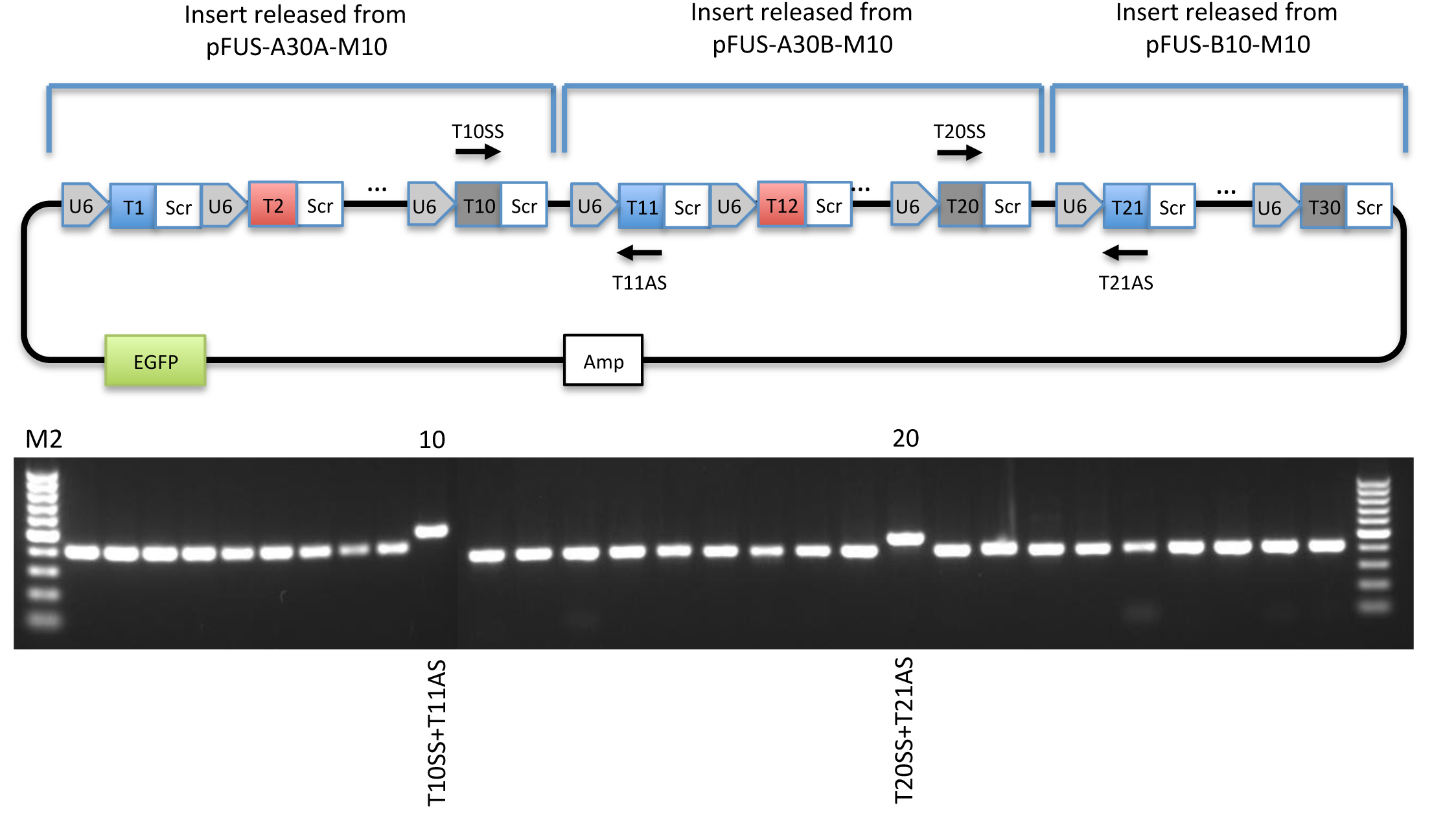
Figure 5. Overlap PCR screening. Top: Schematic illustration of overlap PCR screening of pMsgRNA-EGFP-M30. Bottom: Example of guide specific PCR validation of pMsgRNA-EGFP-M30 visualized on a 1% agarose gel. Lane 10 and lane 20 contain the overlap PCR reactions described above. Note that the overlap PCR produces a slightly larger band than the guide specific PCRs. Generating a theoretical plasmid sequence map is highly recommended as a guideline for assembly, PCR validation, and restriction enzyme digestions. - (Optional) Sanger sequencing using T# as primers.
- Select one positive colony and transfer 50 µl of initial culture to individual tubes containing 5 ml LB medium with 50 µg/ml ampicillin added.
- Incubate overnight at 37 °C in an orbital shaker.
- Inspect plate for positive white colonies. The expected ratio of white to blue colonies is > 80% for successful assembly.
- Day 7. Plasmid DNA preparation and validation
- Isolate the plasmid DNA from the overnight culture using a commercial miniprep kit according to manufacturer’s instructions.
- Validate the assembled pMsgRNA-EGFP-M# array plasmid by restriction enzyme digestion to release the combined gRNA expression array cassette. Add to a PCR tube:
- 0.5 µg assembled array plasmid DNA.
- 1 µl SfiI restriction enzyme.
- 2 µl 10x reaction buffer (to a final concentration of 1x).
- Nuclease-free water to 20 µl total reaction volume.
- Visualize digestion product on a 1% agarose gel by electrophoresis. The expected size of the released expression cassette is approximately 1662 + N x 379 bp (N is the total number of gRNA expression cassettes contained in the final array plasmid).
Notes
- For the efficiency of cloning in each assembly reaction, we generally experience higher than 95% positive rates for the single gRNA expression vectors. This positive rate is based on PCR screening. The positive rate for the assembly of gRNA expression arrays is based on the ratio of white (positive) to blue (negative) colonies. Please note that all blue colonies are negative. However, the white colonies may also be negative if they contain incorrect ligation or recombination products. It is therefore important to perform PCR screenings.
- If a high blue colony rate is observed, it is recommended to check the expiry date of your enzymes as the FastDigest enzymes have a relatively short storage time. Other measures such as lowering the amount of the pFUS plasmid in assembly reactions, increasing cycle numbers, and increasing the amount of enzyme can increase the white colony rate as well.
Acknowledgments
JVN and ALN were supported by grants from the Lundbeck Foundation, Krista og Viggo Petersens Fond, Fabrikant Einar Willumsens Mindelegat, and Fonden til Lægevidenskabens Fremme. LL and LB were supported by the DREAM project from the Lundbeck Foundation. YL was supported by grants from the Danish Research Council for Independent Research, the Sapere Aude Young Research Talent prize to YL, the Lundbeck Foundation and the Innovation Fund Denmark (BrainStem). This protocol is developed based on our previous study published in Cell Mol Life Sci (Vad-Nielsen et al., 2016). We also acknowledge Voytas DF’s group, who constructed plasmids for the first assembly used in this protocol (Cermak et al., 2011).
References
- Cermak, T., Doyle, E. L., Christian, M., Wang, L., Zhang, Y., Schmidt, C., Baller, J. A., Somia, N. V., Bogdanove, A. J. and Voytas, D. F. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39(12): e82.
- Vad-Nielsen, J., Lin, L., Bolund, L., Nielsen, A. L. and Luo, Y. (2016). Golden Gate Assembly of CRISPR gRNA expression array for simultaneously targeting multiple genes. Cell Mol Life Sci 73(22): 4315-4325.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Vad-Nielsen, J., Lin, L., Jensen, K. T., Nielsen, A. L. and Luo, Y. (2016). A Golden Gate-based Protocol for Assembly of Multiplexed gRNA Expression Arrays for CRISPR/Cas9. Bio-protocol 6(23): e2059. DOI: 10.21769/BioProtoc.2059.
Category
Plant Science > Plant molecular biology > DNA > DNA modification
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.