- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Heterologous Expression and Purification of Catalytic Domain of CESA1 from Arabidopsis thaliana
Published: Vol 6, Iss 20, Oct 20, 2016 DOI: 10.21769/BioProtoc.1965 Views: 7553
Reviewed by: Arsalan DaudiSibongile MafuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Heterologous expression of plant cellulose synthase (CESA) and its purification has remained a challenge for decades impeding detailed biophysical, biochemical and structural characterization of this key enzyme. An in-depth knowledge of structure and function of CESA proteins would enable us to better understand the hierarchical structure of the plant cell wall. Here, we report a detailed, and reproducible method of purification of catalytic domain of CESA1 from Arabidopsis thaliana that was recombinantly expressed in Escherichia coli. The method relies on a two stage purification procedure to obtain the catalytic domain in monomer and trimer forms. The biochemical and biophysical data including low resolution structures of the protein have been published (Vandavasi et al., 2016). Currently the crystallization studies of this protein are underway.
[Background] Cellulose is the most important structural component of plant cell walls and constitutes the Earth’s largest source of biorenewable material, yet the mechanism of its synthesis by plants is poorly understood. The plant cellulose synthesis complex (CSC), also called a ‘rosette’ because of its hexameric appearance in electron microscope images, is a large multi-subunit transmembrane protein complex responsible for synthesis of cellulose chains and their assembly into microfibrils. The number of cellulose synthase (CESA) proteins in the CSC and the number of cellulose chains in a microfibril have been debated for many years. Structural information about CESA proteins from plants is crucial to provide answers to some of the basic questions regarding the mechanism of cellulose synthesis. However, elucidation of the structure of CESA proteins has proved difficult because they are multi-domain proteins comprised of disordered, globular, and membrane associated domains. As an alternative to pursuing structural studies of CESA holoproteins, we are developing approaches for recombinant expression of individual CESA domains (e.g., N-terminal domain, central-cytosolic domain, C-terminal transmembrane domain) in large quantities suitable for structural studies. The current protocol has been optimized for isolation of the catalytic domain of A. thaliana CESA1 as reported (Vandavasi et al., 2016). Using this protocol, it is possible to control the oligomerization state of the protein enabling structural studies of the monomer and the trimeric form of the protein. The approach described may be broadly applicable to other systems.
Materials and Reagents
- 0.2 µm filter (VWR, catalog number: 28145-501 )
- E. coli BL21-RIL (Agilent Technologies, catalog number: 230280 )
- Deionized water
- Ampicillin (VWR, catalog number: 97061-442 )
- Chloramphenicol (VWR, catalog number: EM-3130 )
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- Luria broth (EMD Millipore, catalog number: 71751 )
- Sorbitol (Sigma-Aldrich, catalog number: S1876 )
- IPTG (Teknova, catalog number: I3325 )
- Tris buffer (Sigma-Aldrich, catalog number: 252859 )
- Sodium deoxycholic acid (Geno Technology, catalog number: DG090 )
- Nonidet (Sigma-Aldrich, catalog number: I8896 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888 )
- Lysozyme (VWR, catalog number: CA-EM5960 )
- β-mercaptoethanol (BME) (Sigma-Aldrich, catalog number: M6250 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- CAPS buffer (Sigma-Aldrich, catalog number: C2632 )
- Sodium lauroyl sarcosine (Sigma-Aldrich, catalog number: L9150 )
- Glucose (Sigma-Aldrich, catalog number: G5767 )
- Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: D0632 )
- HEPES buffer (Sigma-Aldrich, catalog number: H3375 )
- LB media supplemented with 0.25 M sorbitol (see Recipes)
- Lysis buffer (see Recipes)
- Wash buffer-1 (see Recipes)
- Wash buffer-2 (see Recipes)
- Solubilization buffer (see Recipes)
- Dialysis buffer-1 (see Recipes)
- Dialysis buffer-2 (see Recipes)
Equipment
- Glassware
- Autoclave (Panasonic, model: MLS-3781L )
- Filtering devices for sterilization (Thermo Fisher Scientific, Fisher Scientific, catalog number: 596-4520 )
- Bio hood (Labconco, model: 3440009 )
Note: This product has been discontinued. - Refrigerator or cold room
- Temperature controlled shaker incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: MxQTM 6000 )
- Magnetic stirring plate (VWR, catalog number: 97042-626 )
- UV/Vis spectrophotometer (Thermo Fisher Scientific, Thermo Scientific, model: NanoDrop-2000 )
- Apparatus for SDS-PAGE (Mini-PROTEAN® Tetra vertical electrophoresis cell) (Bio-Rad Laboratories, catalog number: 1658004 )
- 400 Watt sonicator (All-Spec, BRANSON, model: S450D )
- Refrigerated centrifuge (Thermo Fisher Scientific, Thermo ScientificTM, model: RC 6 Plus )
- 10 kDa MWCO PES concentrators (Vivaproducts, model: Vivaspin® 20 )
- Superdex 200 size exclusion chromatographic column, 120 ml capacity (GE Healthcare, catalog number: 17-1069-01 )
Note: This product has been discontinued. - Akta FPLC system (or similar liquid chromatography system)
- Dialysis cassettes (Slide-A-LyzerTM dialysis cassette G2, 10 kDa MWCO) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 87730 )
Procedure
- Cell culture and protein expression
The details of cloning can be found in Vandavasi et al. (2016). A plasmid (pET300NT) containing the gene to express the catalytic domain of CESA1 (amino acids 341-845; MW: 59.7 kDa) from A. thaliana (AtCESA1catD) was transformed into E. coli BL21-RIL strain with ampicillin and chloramphenicol as markers for selection and preserved as a 30% glycerol stock at -80 °C. The plasmid DNA is available on request from Dr. Hugh O’Neill at the Oak Ridge National Laboratory.- Inoculate a starter culture of 50 ml of LB media supplemented with ampicillin (100 µg/ml) and chloramphenicol (35 µg/ml) with a dab of the frozen glycerol stock and grow overnight at 37 °C in a shaking incubator with a shaking speed of 250 rpm.
- Transfer 25 ml of starter culture to 1 L of LB media supplemented with 0.25 M sorbitol, ampicillin (100 µg/ml) and chloramphenicol (35 µg/ml).
- Grow the 1 L culture at 37 °C until the optical density measured at 600 nm in a spectrophotometer (OD600nm) reaches 0.3-0.4 (approximately 2.5 h of growth).
- Change the temperature of incubator to 16 °C and continue cell growth for 15-20 min and monitor the OD600nm. When the OD600nm reaches 0.5-0.6 remove 1 ml of culture (pre-induction sample) for SDS-PAGE analysis.
- When OD600nm is 0.5-0.6 add IPTG to the culture to a final concentration of 1 mM to induce the expression of the AtCESA1catD.
- Incubate the culture at 16 °C for 10-12 h. Remove 0.25 ml of cells (post induction sample) for SDS-PAGE analysis.
- Harvest the cells by centrifuging at 4 °C and 7,000 x g for 15 min and store the cell pellet at -80 °C.
- Analyze the expression levels of AtCESA1catD in pre and post induction samples using SDS-PAGE (Figure 1).
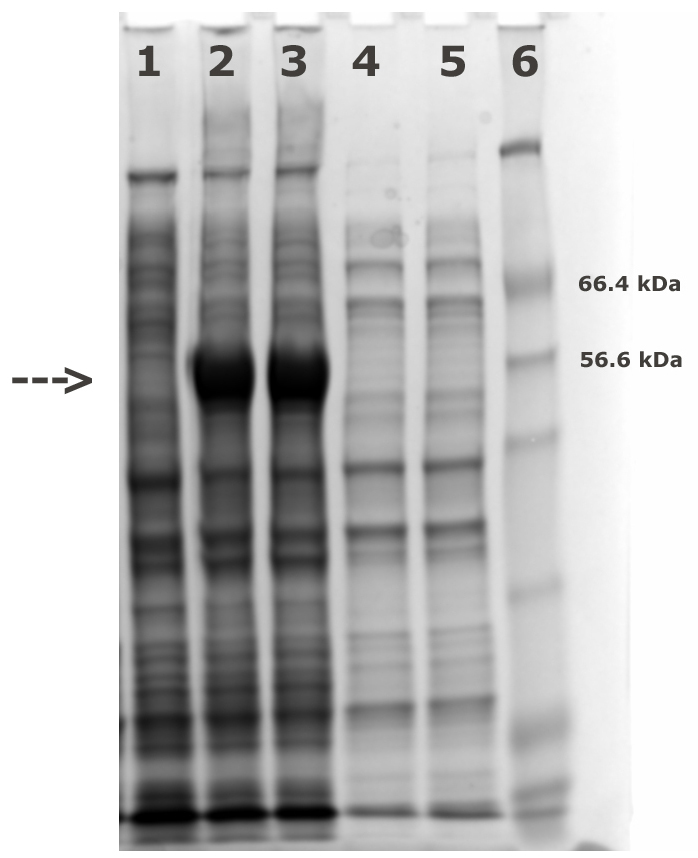
Figure 1. SDS-PAGE analysis of pre and post induction. Lane 1: pre-induced whole cells; Lanes 2, 3: post-induced whole cells; Lanes 4, 5: soluble fraction from post-induced cells; Lane 6: Mw markers. The intense bands at approximately 59 kDa marked with an arrow in lanes 2 and 3 compared to the lane 1 represent induction of the AtCESA1catD protein.
- Inoculate a starter culture of 50 ml of LB media supplemented with ampicillin (100 µg/ml) and chloramphenicol (35 µg/ml) with a dab of the frozen glycerol stock and grow overnight at 37 °C in a shaking incubator with a shaking speed of 250 rpm.
- Lysis, extraction and purification of inclusion bodies
- Resuspend the cell pellet in lysis buffer. 5 ml of lysis buffer per gram of cells (wet weight) worked best for us.
- Incubate the cells in lysis buffer for 30 min on ice. Mix the contents using a pipet occasionally.
- Sonicate the cell suspension. We applied 6 pulses, each for 30 sec, using a 1.3 cm (diameter) horn and 400 Watt sonicator at 50% power. A cooling period of 60 sec was included between consecutive pulses to avoid heating of the sample.
- Centrifuge the cell lysate at 36,000 x g for 30 min at 4 °C to separate the supernatant from the insoluble fraction.
- Discard the supernatant and wash the insoluble fraction with 20 ml of wash buffer-1. The washing involves sequential resuspension of the insoluble fraction using a pipette in 20 ml of wash buffer-1 followed by centrifugation at 36,000 x g for 30 min. Repeat the wash for 3 times.
- Now wash the insoluble fraction with wash buffer-2 for three times.
Note: This step is very important and can be optimized by monitoring the removal of impurities in the insoluble fraction using SDS-PAGE. - At this stage the insoluble fraction contains inclusion bodies that can be dissolved in the solubilization buffer or can be stored at -80 °C for long term storage. The inclusion bodies contain approximately 100 mg of protein per liter of culture and so it is suggested to divide the protein into smaller aliquots before storing.
- Resuspend the cell pellet in lysis buffer. 5 ml of lysis buffer per gram of cells (wet weight) worked best for us.
- Solubilization of inclusion bodies and size exclusion chromatography at 4 °C
- Solubilize the inclusion bodies obtained from 1 L culture in 10 ml of ice cold solubilization buffer by gentle pipetting and incubation for 20-30 min on ice with gentle mixing.
- After 30 min centrifuge the entire contents at 16,000 x g for 45 min to remove insoluble impurities. Collect the supernatant and check the absorbance at 280 nm to estimate the protein concentration. The extinction coefficient of AtCESA1catD is 80,510 M-1 cm-1.
- The final protein concentration of the solubilized protein should be in the range of 15-20 mg/ml. A high concentration of sample is a prerequisite to achieve an efficient separation using size exclusion chromatography. Concentrate the sample to 15-20 mg/ml if necessary using 10 kDa MWCO PES concentrators.
- An Akta FPLC chromatography system was used.
- Equilibrate a Superdex 200 (120 ml) column in solubilization buffer.
- Filter the solubilized sample through 0.2 µm membrane filters and load 1-2 ml of 15-20 mg/ml of sample on to the Superdex 200 (120 ml) column. Set the flow rate at 1 ml/min. In size exclusion chromatography, the sample volume is critical for optimizing the separation of proteins. For a 120 ml column, the optimal sample volume is in the range of 1-2 ml. The combination of a relatively high concentration of sample (15-20 mg/ml) and a small sample volume resulted in a resolved peak of the target protein at a relatively high concentration.
- Collect the fractions (1 ml) for analysis using SDS-PAGE to judge the purity (see Figures 2 and 3). Pool the fractions with pure AtCESA1and concentrate to 10-15 mg/ml using a 10 KDa MWCO PES concentrator. For example, Lanes 3-7 of the SDS-PAGE gel in Figure 3 represent the main peak fractions of the elution profile in Figure 2. These are the fractions that were pooled for further study.
- Dialyze the concentrated and pure sample (1-5 ml) against 1 L of dialysis buffer-1 for 24 h to obtain the pure catalytic domain in monomeric form.
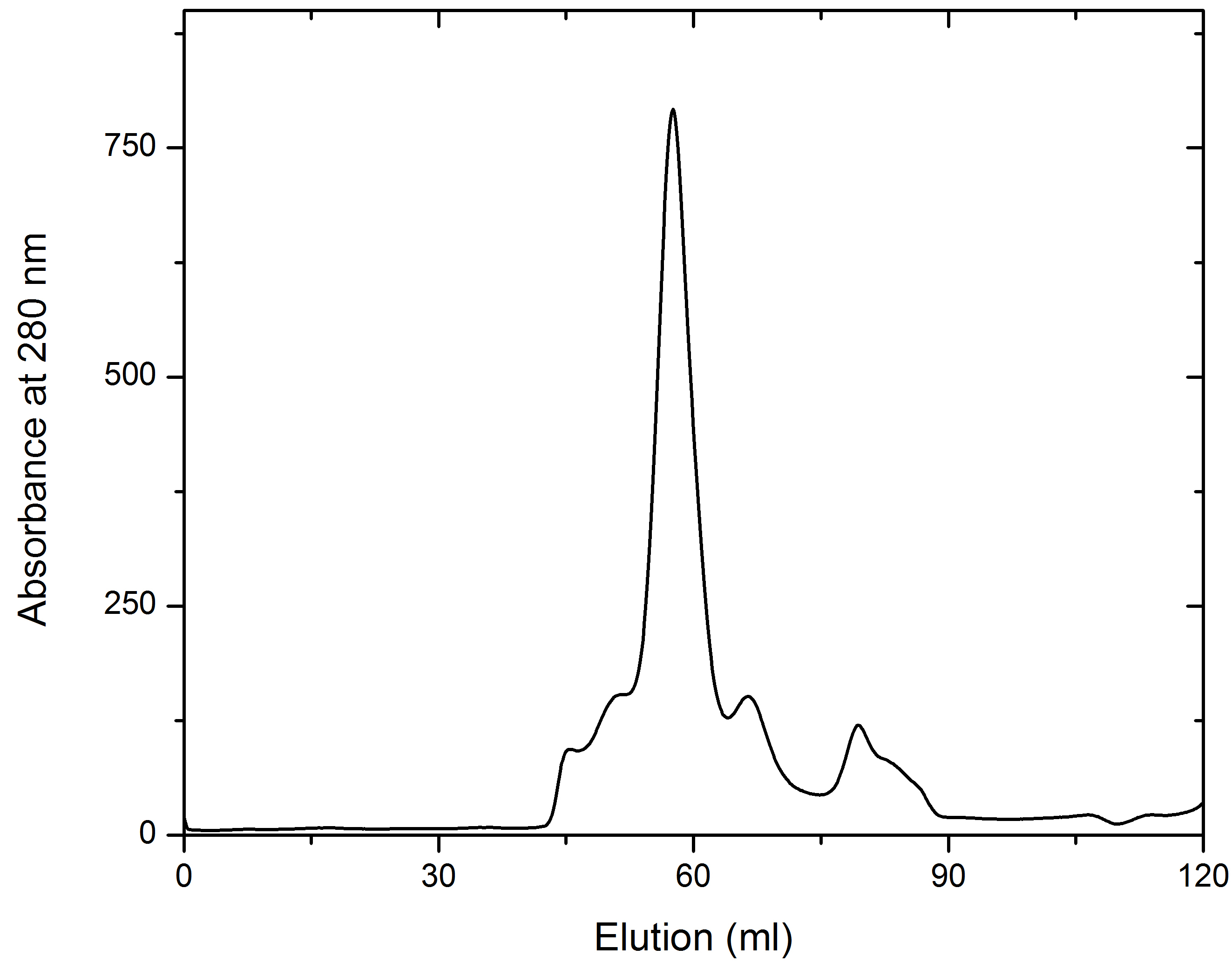
Figure 2. Representative elution profile of AtCESA1catD through a Superdex 200 (120 ml) chromatographic column. 1.5 ml of sample was loaded. Fractions were of 1 ml and the flow rate was set at 1 ml/min. Absorbance is expressed in milli-absorbance units.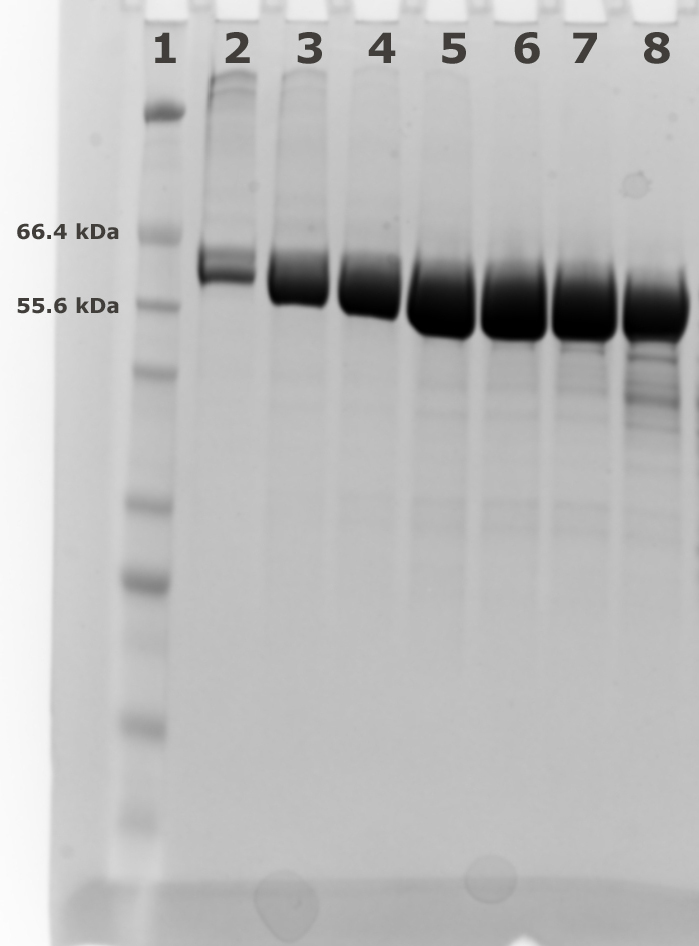
Figure 3. Fractions from Superdex 200 (120 ml) size exclusion chromatography. Lane 1: Marker; Lanes 2-8: Fractions 53 to 59 from size exclusion chromatography.
- Solubilize the inclusion bodies obtained from 1 L culture in 10 ml of ice cold solubilization buffer by gentle pipetting and incubation for 20-30 min on ice with gentle mixing.
- Oligomerization of monomers into trimers at 4 °C
- The monomeric protein self-assembles into trimer by gradual removal of sodium lauroyl sarcosine during dialysis. Typically, 1-2 ml of monomeric protein at a protein concentration of 10-15 mg/ml is dialyzed against 1 L of dialysis buffer-2 for 48 h at 4 °C. The oligomerization of the monomeric protein into a trimer can be monitored by measuring the hydrodynamic radius of the sample using dynamic light scattering (DLS) (Vandavasi et al., 2016).
Data analysis
No special data processing or analysis was required for this procedure.
Notes
- IPTG induction: When OD600nm is 0.5-0.6 add IPTG to the culture to a final concentration of 1 mM to induce the expression of the AtCESA1catD. Based on our experience, this step is very important because we have found that the protein expression is not induced if the OD600nm exceeds 0.8.
- Protein concentration: We suggest using concentrators with PES (polyether sulfone) membranes to concentrate the CESA protein because carbohydrate binding proteins can bind to concentrators that have regenerated cellulose membranes.
Recipes
- LB media supplemented with 0.25 M sorbitol
Dissolve 20 g of LB (peptone from casein: 10 g; yeast extract: 5 g; NaCl: 5 g) in 750 ml of ultrapure water and sterilize by autoclaving.
Prepare 250 ml of 1 M sorbitol and sterilize by filtering through a 0.2 µm filter.
Mix both LB and sorbitol under aseptic conditions. - Lysis buffer
20 mM Tris-HCl, pH 8.0
200 mM NaCl
0.2 mg/ml lysozyme - Wash buffer-1
20 mM Tris, pH 8.0
1% sodium deoxycholic acid
1% Nonidet
10 mM BME - Wash buffer-2
20 mM Tris, pH 8.0
1% Triton X-100 - Solubilization buffer
50 mM CAPS (pH 10.5)
0.2% (w/v) sodium lauroyl sarcosine
0.2 M glucose
3% (v/v) glycerol
5 mM DTT
Note: The pH of the solubilization buffer should be 10.5 measured at room temperature (25 °C). We found that at pH < 10.5 the protein is not readily soluble. - Dialysis buffer-1
50 mM CAPS, pH 8.0
0.2% (w/v) sodium lauroyl sarcosine
0.2 M glucose
3% (v/v) glycerol
5 mM DTT - Dialysis buffer-2
20 mM HEPES, pH 8.0
50 mM NaCl
10% glycerol
5 mM DTT
Notes:
a. NaOH was used to adjust the pH of all the buffers used in this protocol.
b. All the buffers were sterile filtered using 0.2 µm filters.
Acknowledgments
This work was supported by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory (ORNL), managed by UT-Batelle, LLC, for the U. S. Department of Energy (DOE) under contract No.DE-AC05-00OR22725 and by the Center for Lignocellulose Structure and formation (CLSF), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences.
This manuscript has been authored by UT-Battelle, LLC under Contract No.DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
References
- Vandavasi, V. G., Putnam, D. K., Zhang, Q., Petridis, L., Heller, W. T., Nixon, B. T., Haigler, C. H., Kalluri, U., Coates, L., Langan, P., Smith, J. C., Meiler, J. and O'Neill, H. (2016). A structural study of CESA1 catalytic domain of Arabidopsis cellulose synthesis complex: evidence for CESA trimers. Plant Physiol 170(1): 123-135.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Vandavasi, V. G. and O’ Neill, H. (2016). Heterologous Expression and Purification of Catalytic Domain of CESA1 from Arabidopsis thaliana. Bio-protocol 6(20): e1965. DOI: 10.21769/BioProtoc.1965.
- Vandavasi, V. G., Putnam, D. K., Zhang, Q., Petridis, L., Heller, W. T., Nixon, B. T., Haigler, C. H., Kalluri, U., Coates, L., Langan, P., Smith, J. C., Meiler, J. and O'Neill, H. (2016). A structural study of CESA1 catalytic domain of Arabidopsis cellulose synthesis complex: evidence for CESA trimers. Plant Physiol 170(1): 123-135.
Category
Plant Science > Plant biochemistry > Protein > Structure
Plant Science > Plant biochemistry > Protein > Isolation and purification
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.