- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cytology and Microscopy: Immunolocalization of Covalently Modified Histone Marks on Barley Mitotic Chromosomes



Published: Vol 6, Iss 12, Jun 20, 2016 DOI: 10.21769/BioProtoc.1841 Views: 9649
Reviewed by: Marisa RosaXinyan ZhangYurong Xie
Original research article
The authors used this protocol in:

Oct 2015
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Barley is a diploid inbreeding crop with a genome of 5 GB organized into seven chromosomes. The relatively low chromosome number and their large size make barley an excellent model for chromosome cytogenetic studies of large genome cereal crops. Chromatin can be defined as euchromatin or heterochromatin. Euchromatin is gene-rich, less condensed, and transcriptionally active while the heterochromatin is gene-poor, remains highly condensed and has low transcriptional activity (Bartova et al., 2008; Sharakhov and Sharakhova, 2015). However, the mapping of nine Histone modifications has shown that this simple description is not accurate in barley. Instead, it has been shown that combinations of histones carrying different covalent modifications reveal 10 chromatin states partitioning barley chromosomes into three global environments (Baker et al., 2015). Briefly, in this protocol, barley roots (cv Morex) were collected, fixed in paraformaldehyde and squashed onto slides. Chromosome spreads were immunostained using antibodies against specific histone modifications, in particular H3K27me3, K3K27me1 and H3K9me2. We used confocal imaging to acquire stacked images and confirm the locations of these histone modifications on barley chromosomes.
Keywords: BarleyMaterials and Reagents
- SterilinTM 9 cm petri dish
(Thermo Fisher Scientific, catalog number: 1011RR )
- Whatman® 9 cm filter paper (Sigma-Aldrich, catalog number: Z240079 )
- Fine marker pen permanent (STABILO International GmbH)
- HB graphite pencil
- 15 ml centrifuge tube (any brand)
- Single edge razor blade (Thermo Fisher Scientific, catalog number: 11904325 )
- 1.5 ml microcentrifuge tubes
(VWR International, catalog number: 89000-028 )
- Microscope Polysine® slide (Thermo Fisher Scientific, Menzel Glaser, catalog number: 10143265 ) or Superfrost® plus micro slide (VWR International, catalog number: 48311-703 )
Note: Currently, it is “Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 10143265 ”. - Borosilicate glass coverslip, 25 x 50, thickness 0.13 mm (VWR International, catalog number: 631-0137 )
- Tissue paper
- PAP pen for immunostaining (Sigma-Aldrich, catalog number: Z377821 )
- 0.45 μm, 26 mm diameter syringe filter (Cole-Parmer, catalog number: UY-02915-58 )
- Glass Coplin staining jar (any brand)
- Seeds from barley cv. Morex
- Immersion oil, ImmersolTM W 2010 (Carl Zeiss)
- Autoclaved distilled water (Barnstead® E-pure 4 modules cartridges)
- 5% bleach (Domestos)
- 70% ethanol
- Liquid nitrogen
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A7030 )
- Antibodies
- Primary antibodies: anti-trimethyl-histone H3 (Lys27) antibody (H3K27me3) (Merck Millipore Corporation, catalog number: 07-449 ), anti-monomethyl-histone H3 (Lys27) antibody (K3K27me1) (Merck Millipore Corporation, catalog number: 07-448 ) and histone H3K9me2 antibody (pAb) (H3K9me2) (Active Motif, catalog number: 39375 )
Note: Currently, it is “EMD Millipore Corporation, catalog numbers: 07-449 and 07-448 ”. - Secondary antibodies: goat anti-Rabbit IgG (H+L), Alexa Fluor® 488 conjugate (Thermo Fisher Scientific, catalog number: A-11034 )
- Primary antibodies: anti-trimethyl-histone H3 (Lys27) antibody (H3K27me3) (Merck Millipore Corporation, catalog number: 07-449 ), anti-monomethyl-histone H3 (Lys27) antibody (K3K27me1) (Merck Millipore Corporation, catalog number: 07-448 ) and histone H3K9me2 antibody (pAb) (H3K9me2) (Active Motif, catalog number: 39375 )
- Hoechst 33342 (10 mg/ml) (Thermo Fisher Scientific, Molecular ProbesTM, catalog number: H3570 )
- Vectashield antifade mounting medium (Vector Laboratories, catalog number: H-1000 )
- Clear nail varnish (Technic)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: 255793 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5379 )
- Tween® 20 (Sigma-Aldrich, catalog number: P9416 )
- TritonTM X-100 (Sigma-Aldrich, catalog number: T8787 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: S8045 )
- Paraformaldehyde (Sigma-Aldrich, catalog number: P6148 )
- Cellulase Onozuka R10 (Duchefa Biochemie, catalog number: C8001 )
- Pectolyase Y23 (Dushefa Biochemie, catalog number: P8004 )
- 10x PBS (see Recipes)
- 1x PBS, 0.5% Triton® X-100 (see Recipes)
- 1 M NaOH (see Recipes)
- 4% formaldehyde (see Recipes)
- 0.1 M citric acid (see Recipes)
- 0.1 M sodium citrate (see Recipes)
- Citrate buffer (see Recipes)
- Enzyme mixture (see Recipes)
- Blocking solution (see Recipes)
Equipment
- Optoelectronically controlled small orbital shaker, 2,200 rpm, 230 V (IKA, model: VXR basic Vibrax® )
- Stereomicroscope (optional) (TEC Microscopes LTD., model: HM-4 )
- Light microscope (TEC Microscopes LTD., model: LM-2TR )
- Confocal microscope LSM 710 (Carl Zeiss)
- Objective (for Zeiss LSM 710): 60x APO C-apochromat 63x/1.20 W Korr M27
- DAPI fluorescence (blue 405, UV lamp) (Hoechst)
- Fitted 405 nm diode laser (for Hoechst acquisition on the LSM 710)
- Fitted 488 argon laser (for Alexa Fluor® 488 acquisition on the LSM 710)
- Moisture or wet chamber
Note: Commercially available for immunohistochemistry application or home made by using a plastic box lined with wet paper and wax sheet or glass rod to keep the slides out of direct contact with water (Figure 1). The chamber needs to be closed so the humidity is preserved during the protocol. - Aluminium slide tray, 20 slides capacity (Brunel Microscopes Ltd.)

Figure 1. Wet chamber. The principle of the wet chamber is to keep the sample moist at all times.
Software
- Zen 2010 software (Carl Zeiss, version 6.0)
- Fiji (Image J, version 1.49m) (Schindelin et al., 2012) with deconvolution package (EPFL, Biomedical Imaging Group, Switzerland)
- Imaris (optional) (Bitplane AG, version 8.1.2)
Procedure
- Sample preparation
Note: All steps were done at room temperature on the bench (no need for sterile conditions).- Seeds sterilization
- Wash the seeds in 15 ml tube for 15 min in 5% bleach-shaking (5 ml).
- Discard the bleach and rinse 3 times for 5 min in sterile water-shaking (5 ml) in the same tube.
- Discard the water and wash the seeds in 70% ethanol for 5 min (5 ml) in the same tube.
- Discard the Ethanol and rinse 3 times for 5 min in sterile water in the same tube.
- Wash the seeds in 15 ml tube for 15 min in 5% bleach-shaking (5 ml).
- Seeds germination
- Place 4 to 6 seeds per petri dish containing a wet filter paper (sterile water).
- Write the genotype and date on the lid with the fine marker pen.
- Cover the plate with foil.
- Leave at room temperature for one day and let the seeds germinate for about 5 days at 4 °C (Figure 2).
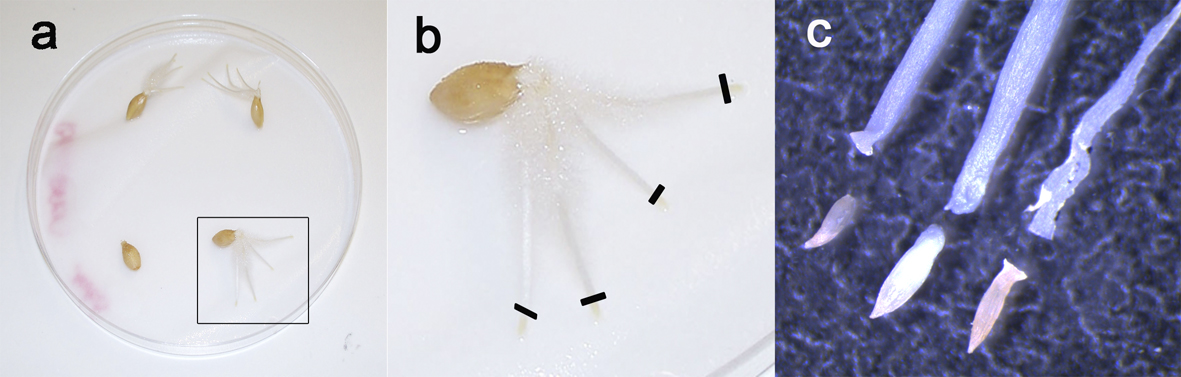
Figure 2. Root tips preparation. Barley seeds after 5 days germination (a) showing 3 to 4 roots. To prepare chromosomes spreads, only the yellow tips are taken (b, black lines), because they contain the dividing tissue while the remaining part of the roots is mainly composed of non-mitotic elongating cells. c. Close-up of the root tips under stereomicroscope.
- Place 4 to 6 seeds per petri dish containing a wet filter paper (sterile water).
- Seeds sterilization
- Slide preparation
- Chromosome spread preparation
- Cut the root tips (Figure 2a) under the stereomicroscope (Figure 2b) to isolate the yellow part (Figure 2c).
- Place in a 1.5 ml tube with 0.5 ml distilled water (dH2O).
- Place the tube on ice for at least 6 h (best overnight or 24 h), no shaking. Cells are arrested in metaphase during 6-24 h, therefore increasing the Mitotic Index Number of metaphase.
- Replace the cold water with fresh 4% formaldehyde (0.5 ml) and fix for 30 min at room temperature, no shaking. Formaldehyde has to be made fresh every time for reliability and using a fume hood (formaldehyde is carcinogenic).
- Rinse 3 x 5 min in 1x PBS (0.5 ml), in the fume hood. Discard in formaldehyde waste according to local laboratory rules.
- Digest roots with the enzyme mixture for 30-45 min at 37 °C. This step can be done in the 1.5 ml tube or any container such as an embryo dish. Root tips should become very soft after digestion. If not then, leave the root tips for further 20 min period(s) up to 2 h total digestion, until the roots can be easily squashed.
- Wash twice for 5 min with cold 1x PBS, 0.5% Triton X-100 at room temperature.
- Place 3 or 4 roots tips on a microscope slide (Figure 3a), add a 22 x 22 coverslip (see orientation on Figure 3b) and squash with your finger gently. Do not twist or roll the thumb from side to side. Be careful when handling the roots.
- Immerse the slide fully in liquid nitrogen using long forceps (wear appropriate safety protection according to the lab regulations). When the slide/coverslip is cooled enough (bubbling stops), remove the coverslip very quickly by inserting the razor blade (Figure 3b, grey shape) under one of the coverslip corners (Figure 3b).
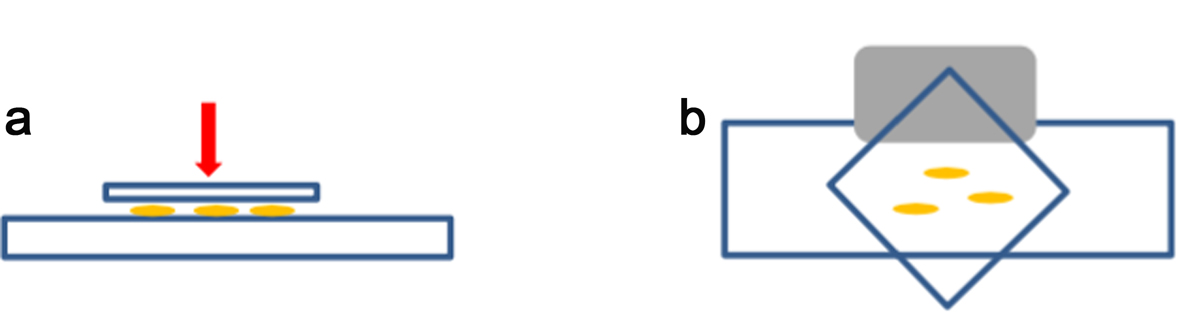
Figure 3. Squash method. For squashing, enzymatically digested root tips are placed between a microscope slide and a coverslip (a). The coverslip is placed as shown in (b) to be removed easily by using the edge of a razor blade (grey shape) under one side of the coverslip after freezing in liquid nitrogen. - Lay the slide on an aluminium tray and let it air dry gently (see Note 1). Check the spread quality by looking at the chromosomes spread under a light microscope (using a 40x objective at least). Drying time normally takes no longer than 5-15 min at room temperature (21 °C), but this will depend on the temperature and humidity of the lab. It is best to process the slides as soon as they look dry.
Note: Never use high temperatures as drying the slides too harshly will increase the background during immunostaining. Examine the samples quickly under the microscope. If necessary, add a drop of PBS onto the slide to check your spreads. For better results, use freshly made slides. Slides can be stored in the freezer for up to a month, but this will affect reproducibility. Leave the slide to dry for 5-15 min (depending on the lab environmental conditions). Add a drop of distilled water or PBS to the samples to keep them wet prior to adding the primary antibody. - Use the slides immediately.
Note: Room temperature equals 21 °C, reproducibility of the protocol may vary if the temperature is higher than 21 °C, mainly by drying the sample too quickly.
- Cut the root tips (Figure 2a) under the stereomicroscope (Figure 2b) to isolate the yellow part (Figure 2c).
- Chromosome spread preparation
- Immunostaining
- Use the PAP pen to circle the sample and let it dry.
Note: It is possible to leave the slide for longer than 90 min, but longer exposure to the secondary antibody may increase the slide background with unbound antibody. - Optional - Block the sample with the blocking solution for 20 min at room temperature in the wet chamber. This step did not make any difference for the antibodies used in this study but could improve the protocol for the use of other antibodies not yet tested.
- Apply 30 μl of primary antibody solution to the center of this circle.
Note: Primary antibody solution consists of 1 μl of histone antibody in 100 μl of blocking solution. - Cover with a “soft” coverslip of Parafilm M® or cut autoclave bag. The dimension of these coverslips should match the slide dimensions (approximatively 22 x 50 mm).
- Place the slide in a wet chamber (see Figure1) for 30 min to 1 h at room temperature (see Note 4).
Notes:- The wet chamber comprises a plastic box lined with wet paper (Figure 1). The slide is placed on wax paper or glass rod, making sure it does not touch the wet surface.
- 10-15 min is normally enough to stain the cells, but this steps can be longer. However, this may increase unspecific background in the blue channel.
- The wet chamber comprises a plastic box lined with wet paper (Figure 1). The slide is placed on wax paper or glass rod, making sure it does not touch the wet surface.
- Incubate slides overnight (and up to 48 h) in the fridge at 4 °C. Remove the slides from the fridge and leave them to warm up for at least 30 min at room temperature.
- Wash the slides in 1x PBS, 0.5% Triton X-100 for at least 15 min (once 15 min or 3 x 5 min), in a glass Coplin staining jar, shaking at 100 rpm.
- Add 30 µl of the secondary antibody and repeat steps C3-4.
- Secondary antibody solution consisted of 1 μl of anti-rabbit Alexa Fluor® 488 in 600 μl of blocking solution.
- Keep all steps in the dark from this point.
- Secondary antibody solution consisted of 1 μl of anti-rabbit Alexa Fluor® 488 in 600 μl of blocking solution.
- Incubate 60-90 min at room temperature and repeat step C7.
Note: Slides mounted in Vectashield and sealed with nail varnish can be kept for months if stored in a dry place at 4 °C. - Remove the excess buffer by keeping the slides vertically on a tissue paper.
- Add 20 μl of 2 μg/ml Hoechst 33342 and incubate for 10-15 min at room temperature.
Note: This step (step C11) can be skipped by using Vectashield already containing DNA staining. Other anti-fade solution could also be used although users should check the solution refracting index (1.45 for Vectashield) that can affect imaging. - Remove the excess stain by placing the slide vertically on tissue paper for 5 sec and add one drop of Vectashield.
- Carefully add a 22 x 50 coverslip, making sure that there are no bubbles underneath.
- Remove the excess of Vectashield by blotting the slide upside down on a tissue paper and seal with the nail varnish.
- Let it dry for at least 15 min and keep the slides in the dark and in the fridge until imaging.
- Use the PAP pen to circle the sample and let it dry.
- Imaging
3D Confocal stack images (Figure 4a-c) were acquired with LSM-Zeiss 710 instrument and the ZEN software.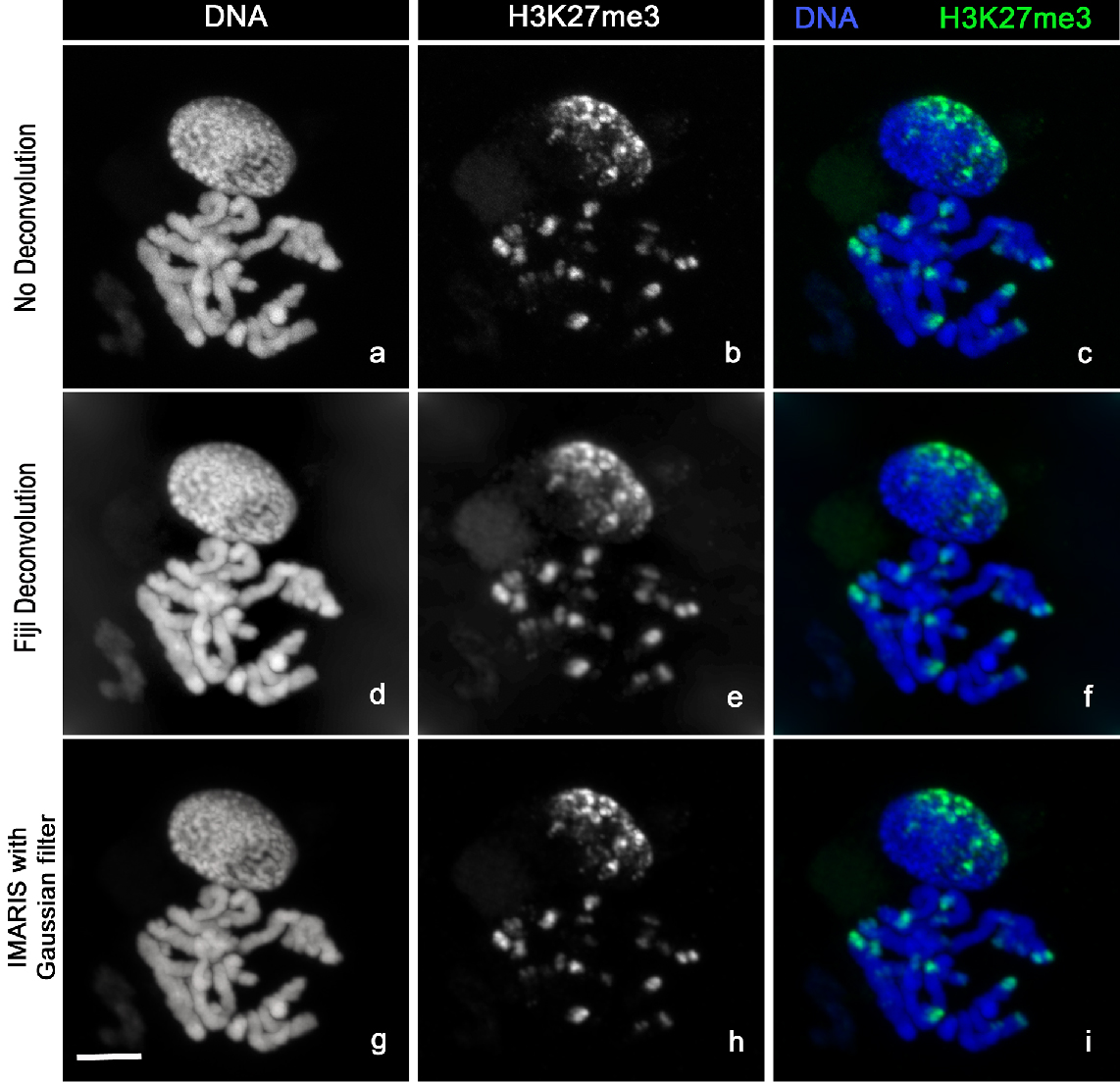
Figure 4. Image analysis. Chromosomes Spread stained with Hoechst 33342 (a, d, g) and H3K27me3-Alexa488 (b, e, h). The images clearly show the distribution of H3K27me3 at the chromosomes ends (c). H3K27me3 modification (c) is mainly localized at one side of the nucleus (n) correlating with the so-called Rabl configuration (telomeres and centromeres are opposite side of the nucleus during Interphase). The sharpness of the raw images (a, b, c) can be improved by using deconvolution with Fiji (d, e, f) or the Imaris Gaussian filter (g, h, i). Scale bar, 5 µm.- Locate your sample under the 10x objective using the DAPI fluorescence then switch to the C-Apochromat 63x/1.20 W Korr M27 objective [use ImmersolTM W (2010)].
- Locate your cell with DAPI and use the function Auto Exposure.
- Zoom on your cell of choice and re-adjust the laser intensity. All images were taken with the following settings:
- Scan mode 0.5 μm stack section
- Pixel Dwell 2.27 μs
- Pinhole 67 μm
- Average line 2
- 12-bits
- Bidirectional
- Scan mode 0.5 μm stack section
- Press run to acquire the image, name and save as .lsm files.
- Locate your sample under the 10x objective using the DAPI fluorescence then switch to the C-Apochromat 63x/1.20 W Korr M27 objective [use ImmersolTM W (2010)].
- Analysis
Images were processed with Fiji (Schindelin et al., 2012) or Imaris 7.7.2 (Figure 3).- Fiji
- In Fiji open your file using Plugins/Bio-Formats/Bio-Formats Importer
- Unclick all option but “Split Channel” and “Autoscale”.
- Select the image of the first channel and open the DeconvolutionLab Plugins (Vonesch and Unser, 2008).
- Choose Tikhonov-Miller algorithm and choose between 2 to 5 iterations. Go to PSF and choose the image to deconvolve. Press run. Another image will be produced. Repeat the same for the second channel.
- Go to Image/Merged Channel and choose your green and blue files names. Tick create composite and keep image, then click ok. You have now a merged 3D stacks.
- Save using the Bio-Formats exporter and choose the .ome extension. This can be opened in many utility software later on.
- Now select your 3D stack, go to Image/Stack/Z Project and choose maximum intensity. Change the colour by using Image/Colour/Channel Tool. Then choose Image/Type/RGB and save your images (Figure 4d-f) with the extension of your choice.
- In Fiji open your file using Plugins/Bio-Formats/Bio-Formats Importer
- IMARIS
- Open your image in Imaris and choose the option “Easy 3D”.
- Choose Image Processing/Smoothing/Gaussian filter for both channels-click OK.
- Click Snapshot, choose “Crop to fill the whole snapshot area”.
- Select the dimension (either your dataset or 512 x 512) and choose the resolution (600 dpi for publication).
- Choose the file location and the file name and click “do Snapshot”.
- Save your file in each channel by selecting/deselecting the channel in the Display Adjustment panels (Figure 4g-i).
- Open your image in Imaris and choose the option “Easy 3D”.
- Fiji
Representative data
Representative data are shown in Figure 4.
Recipes
- 10x PBS
Dissolve the following in 800 ml dH2O
80 g of NaCl
2.0 g of KCl
14.4 g of Na2HPO4
2.4 g of KH2PO4
Adjust pH to 7.4
Adjust volume to 1 L with additional dH2O
Sterilized by autoclaving
Shelf life 6 months
Dilute with dH2O to make 2x PBS or 1x PBS working solution (the shelf life of the diluted solutions is 1 month) - 1x PBS, 0.5% TritonTM X-100
Mix the following
1 L of 1x PBS
5 ml of Triton X-100
Gently warm up the solution at 37 °C for 5 min to help dissolve Triton X-100
Shelf life 1 month - 1 M NaOH
Dissolve 40 g of NaOH in 500 ml of dH2O
Shelf life 6 months - 4% formaldehyde
For 25 ml of 4% formaldehyde solution (under a fume hood)
Dissolve 1 g paraformaldehyde in 12.5 ml dH2O in a glass beaker
Add one or two drop(s) of 1 M NaOH and seal the beaker with foil
Heat at 60 °C at least 5-10 min, the solution must be transparent (do not boil)
Let it cool on ice until you can hold with your hand without burning
Add 12.5 ml of 2x PBS
The solution has to be fresh for best reproducibility - 0.1 M citric acid
Dissolve 2.1 g citric acid in 100 ml dH2O
Autoclaved and stored in the fridge
Shelf life 6 months (1 year if stored at 4 °C) - 0.1 M sodium citrate
Dissolve 2.94 g sodium citrate in 100 ml dH2O
Autoclaved and stored in the fridge
Shelf life 6 months (1 year if stored at 4 °C) - Citrate buffer
Make it freshly by mixing the following:
444 μl of 0.1 M sodium citrate (2x)
556 μl of citric acid (2x)
Adjust to 10 ml with dH2O
Best when freshly made but could also be stored in the fridge at 4 °C for 2 weeks - Enzyme mixture
Dissolve both enzymes in 10 ml citrate buffer:
100 mg of Cellulase Onozuka R10
100 mg of Pectolyase Y23
Aliquot out in 333 lots (optional: Filter through a 0.45 μm filter before)
Stored in freezer up to 1 year at -20 °C
Thaw one aliquot and dilute with 667 μl citrate buffer to make 1 ml working solution
Always made fresh and used on the same day - Blocking solution
Dissolve 0.15 g of BSA in 3 ml 1x PBS, 0.5% Triton X-100
Always made fresh and used on the same day
Acknowledgments
We gratefully acknowledge the financial support from the grant BBSRC BB/I1022899/1 “The diversity and evolution of the gene component of the barley pericentromeric heterochromatin”, the European Community Grant FP7 MeioSys (222883), ERC project 669182 ‘SHUFFLE’ and the Scottish Government through RESAS work programme (WP5.2).
References
- Baker, K., Dhillon, T., Colas, I., Cook, N., Milne, I., Milne, L., Bayer, M. and Flavell, A. J. (2015). Chromatin state analysis of the barley epigenome reveals a higher-order structure defined by H3K27me1 and H3K27me3 abundance. Plant J 84(1): 111-124.
- Bartova, E., Krejci, J., Harnicarova, A., Galiova, G. and Kozubek, S. (2008). Histone modifications and nuclear architecture: a review. J Histochem Cytochem 56(8): 711-72
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Sharakhov, I. V. and Sharakhova, M. V. (2015). Heterochromatin, histone modifications, and nuclear architecture in disease vectors. Curr Opin Insect Sci 10: 110-117.
- Vonesch, C. and Unser, M. (2008). A fast thresholded landweber algorithm for wavelet-regularized multidimensional deconvolution. IEEE Trans Image Process 17(4): 539-549.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Colas, I., Baker, K. and Flavell, A. J. (2016). Cytology and Microscopy: Immunolocalization of Covalently Modified Histone Marks on Barley Mitotic Chromosomes. Bio-protocol 6(12): e1841. DOI: 10.21769/BioProtoc.1841.
Category
Plant Science > Plant molecular biology > Protein
Plant Science > Plant cell biology > Cell imaging
Molecular Biology > Protein > Detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.