- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Terminal Restriction Fragments (TRF) Method to Analyze Telomere Lengths




Published: Vol 5, Iss 23, Dec 5, 2015 DOI: 10.21769/BioProtoc.1671 Views: 20039
Reviewed by: Tie LiuIgor Cesarino Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Chromosome ends - telomeres - are a focus of intensive research due to their importance for the maintenance of chromosome stability. Their shortening due to incomplete replication functions as a molecular clock counting the number of cell divisions, and ultimately results in cell-cycle arrest and cellular senescence. Determination of telomere lengths is an essential approach in telomere biology for research and diagnostic applications. Terminal Restriction Fragments (TRF) analysis is the oldest approach to analyze telomere lengths and remains the “gold standard” even in current studies. This technique relies on the fact that repeated minisatellite telomeric units do not contain target sites for restriction enzymes. Consequently, telomeres remain in relatively long fragments (TRF), whereas the genomic DNA is digested into short pieces. Fragments of telomeric DNA are then visualized by hybridization with radioactively labeled telomeric probe. As TRF include besides telomeres also a short region of telomere-associated DNA up to the first restriction site, results are slightly shifted towards higher TRFs values. Therefore, the use of frequent cutters or their mixtures is recommended to minimize this difference. Moreover, by using TRF analysis it is possible to distinguish genuine (terminal) telomeres from interstitial telomeric repeats (ITR) (Richards and Ausubel, 1988). In this approach, BAL31 digestion is first applied on high molecular weight DNA. The enzyme progressively degrades linear DNA from its ends. The degraded DNA is then digested with one or more restriction enzymes and fragments are separated by gel electrophoresis. After blotting, membranes are probed with either a terminal marker sequence or telomeric sequence. Genuine TRF can be distinguished from ITR due to their progressive shortening with increasing BAL31 digestion time, while ITR are BAL31-resistant. The TRF BAL31 digestion pattern at the time zero indicates the approximate telomere lengths (Fajkus et al., 2005).
Keywords: TelomereMaterials and Reagents
- DNA isolated from plant or animal tissues (Note 1)
- Plug molds (Bio-Rad Laboratories, catalog number: 170-3713 )
- Restriction enzymes-Frequent-cutters restriction enzymes e.g. MseI (New England Biolabs, catalog number: R0525L ), AluI (New England Biolabs, catalog number: R0137S ), BstNI (New England Biolabs, catalog number: R0168S ), HaeIII (New England Biolabs, catalog number: R0108S ) , HinfI (New England Biolabs, catalog number: R0155L ), RsaI (New England Biolabs, catalog number: R0167S )
Note: Reaction buffers are supplied by the manufacturer. - BAL31 nuclease (New England Biolabs, catalog number: M0213L )
- 50 mM Ethylene glycol bis(2-aminoethylether)-N, N, N', N'-tetra acetic acid (EGTA) (pH 8.0) (SERVA Electrophoresis GmbH, catalog number: 11 290.02 )
- 0.5 M EDTA (pH 8.0) (Duchefa Biochemie, catalog number: E0511 )
- Agarose for electrophoresis (SERVA Electrophoresis GmbH, catalog number: 11404.05 )
- Agarose for pulsed field gel electrophoresis (PFGE) (Bio-Rad Laboratories, catalog number: 162-0138 )
- Low melt agarose (Bio-Rad Laboratories, catalog number: 161-3112 )
- Ethidium bromide aqueous solution 1% w/v (10 mg/ml) (SERVA Electrophoresis GmbH, catalog number: 21 251.01 )
- DNA loading buffer (6x concentrated) (Thermo Fisher Scientific, catalog number: R0611 )
- DNA marker for conventional agarose electrophoresis (e.g. GeneRuler 1 kb DNA Ladder) (Thermo Fisher Scientific, catalog number: SM0311 )
- DNA marker for PFGE [e.g. Mid Range or Low Range PFG Marker (New England Biolabs, catalog number: N3551S or N0350S )]
- 0.25 M HCl (Penta Technologies, catalog number: 77232 )
- 0.4 M NaOH (Penta Technologies, catalog number: 71691 )
- 100 mM PMSF (in isopropanol) (SERVA Electrophoresis GmbH, catalog number: 32395 )
- Proteinase K from Tritirachium album min. 8 DMC-U/mg (SERVA Electrophoresis GmbH, catalog number: 33752.02 )
- N-Lauroylsarcosine sodium salt (Sigma-Aldrich, catalog number: L5125 )
- D-mannitol (Duchefa, catalog number: M0803 )
- Nylon membrane (Hybond XL) (GE Healthcare, catalog number: RPN303 S )
- T4 polynucleotide kinase (New England Biolabs, catalog number: M0201L )
- DecaLabel DNA Labeling kit (Life Technologies, catalog number: K0622 )
Note: Currently, it is “Thermo Fisher Scientific, catalog number: K0622”. - 32P-γ-ATP [e.g., Institute of isotopes (Hungary, catalog number: FP-501 )]
- 32P-α-dATP (or 32P-α-dCTP) [Institute of isotopes (Hungary, catalog number: FP-203 )]
- Synthetic telomeric oligonucleotide (4 telomeric repeats (CCCTAAA)4 or (TTTAGGG)4), or telomeric concatemers
- NaCl
- Tris-HCl (pH 8)
- MgCl2
- CaCl2
- Glacial acetic acid
- Boric acid
- NaH2PO4
- Na2HPO4
- SDS
- SSC
- BAL31 nuclease buffer (see Recipes) or can be purchased (New England Biolabs, catalog number: B0213S )
- 50x TAE (see Recipes)
- 5x TBE (see Recipes)
- Hybridization buffer (see Recipes)
- Washing solution (see Recipes)
- TE buffer (see Recipes)
- TEM buffer (see Recipes)
- Proteinase buffer (see Recipes)
Equipment
- Apparatus for standard agarose gel electrophoresis (e.g. Bio-Rad Laboratories, AbD Serotec®)
- PFGE electrophoresis apparatus; for optimal resolution resultion, use system with hexagonal electrodes (e.g. CHEF DR, Bio-Rad Laboratories, AbD Serotec® or Gene Navigator, Amersham)
- Thermo block (e.g. Eppendorf, model: Thermomixer )
- Vacuum concentrator (e.g. Thermo Fisher Scientific, model: SpeedVac )
- Vacuum blotter (e.g. Bio-Rad Laboratories, AbD Serotec®)
- Gel documentation system (e.g. R&D Systems, FujiFilm, model: LAS3000 )
- Hybridization oven [e.g. HybriLinker (Analytik Jena, model: UVP )]
- Phosphoimager (e.g. GE Healthcare Dharmacon, model: FLA7000 )
Software
- Multi Gauge signal processing software (FujiFilm)
Procedure
- Restriction endonuclease(s) digestion
Based on the expected telomere length, either DNA isolated by standard approaches (telomeres ˂ 10 kb) or high molecular weight DNA (telomeres > 10 kb) is digested.- Digestion of genomic DNA isolated by a standard protocol
- Choose appropriate restriction endonuclease or mix of enzymes. These should (i) be frequently cutting with no recognition site inside the canonical telomeric repeat, (ii) cleave under similar reaction conditions (buffer, temperature optimum), (iii) show relatively long survival at working temperature, and (iv) be not sensitive to cytosine methylation.
Examples of restriction enzymes and experimental conditions: MseI, 37 °C; AluI, 37 °C; HinfI + HaeIII, 37 °C; HinfI + RsaI, 37 °C; BstNI, 60 °C. - Digest ~1 μg of genomic DNA with 20 U of enzyme (or mix of enzymes) for 3 h. Add next aliquot of enzyme(s) and digest O/N.
- If necessary for loading the sample on the agarose gel, reduce the volume of reaction mixture using the vacuum concentrator.
- Choose appropriate restriction endonuclease or mix of enzymes. These should (i) be frequently cutting with no recognition site inside the canonical telomeric repeat, (ii) cleave under similar reaction conditions (buffer, temperature optimum), (iii) show relatively long survival at working temperature, and (iv) be not sensitive to cytosine methylation.
- Digestion of high molecular weight DNA
- Choose appropriate restriction endonuclease or mix of enzymes.
- Incubate DNA embedded in the agarose plug (Note 1) in 200-300 μl of the corresponding restriction buffer in 2 ml Eppendorf tube for 30 min at the temperature for the digestion (Note 2).
- Replace the restriction buffer by a fresh aliquot (200 μl per 1 agarose block, 300 μl per 2 agarose blocks), add the restriction enzyme(s) (30 U/1 agarose block), and incubate for 3 h. Add next aliquot of enzyme(s) and digest O/N.
- Analyze DNA in agarose plugs by PFGE. Fraction of low molecular weight DNA released in the course of digestion to the reaction buffer can be isolated by phenol-chloroform extraction and analyzed by conventional agarose gel electrophoresis.
- Choose appropriate restriction endonuclease or mix of enzymes.
- Optional: Digestion with BAL31 nuclease prior to restriction enzyme(s) treatment
To determine the terminal position of the putative telomeric sequence, high molecular weight DNA (Note 1) is treated with BAL31 nuclease to degrade terminal sequences. In the TRF analysis, loss of telomere-specific hybridization signal in the course of BAL31 digestion is observed.- Take six or more DNA agarose blocks (based on the number of BAL31 digestion time intervals, e.g., 0, 10, 20, 40, 60, 90 min), put each to a 2 ml Eppendorf tube, add 200 μl of BAL31 reaction buffer, incubate for 30 min at 30 °C.
- Replace the reaction buffer by the fresh aliquot (200 μl), incubate the tube in a thermo block (30 °C), add 3 U of BAL31 nuclease (except of zero-time sample) and digest for the respective time (digestion time e.g., 10, 20, 40, 60, 90 min).
- Stop the reaction by removing the restriction buffer and adding 500 μl of 50 mM EGTA (pH 8).
- Inactivate BAL31 by incubation at 55 °C for 30 min.
- Replace EGTA by 1.5 ml of 0.1x TE buffer, incubate at 4 °C for 15 min. Repeat the washing step at least twice more.
- Continue with digestion by restriction endonuclease (see step A2).
- Take six or more DNA agarose blocks (based on the number of BAL31 digestion time intervals, e.g., 0, 10, 20, 40, 60, 90 min), put each to a 2 ml Eppendorf tube, add 200 μl of BAL31 reaction buffer, incubate for 30 min at 30 °C.
- Digestion of genomic DNA isolated by a standard protocol
- Separation of DNA by agarose gel electrophoresis
- Conventional agarose electrophoresis (separation of terminal fragments of expected lengths ˂ 10 kb)
- Prepare 1% agarose gel in 1x TAE buffer.
- Cool the gel to ~60 °C, add ethidium bromide to a final concentration of 0.2 μg/ml.
- Add DNA loading buffer to DNA samples, load on the gel. Load DNA marker (1 kb DNA ladder).
- Run electrophoresis (1.2-2.6 V/cm), check positions of the DNA marker fragments.
- Document the gel using a documentation system (in the mode for ethidium bromide-excitation at 312 nm). An example of the Arabidopsis thaliana DNA separated using conventional agarose gel electrophoresis is presented in Figure 1A.
- Transfer DNA to the nylon membrane by capillary or vacuum alkali blotting (depurination in 0.25 M HCl, denaturation and transfer in 0.4 M NaOH), hybridize with radioactively labeled telomeric probe (see step C3).
- Prepare 1% agarose gel in 1x TAE buffer.
- Pulsed field gel electrophoresis (separation of terminal restriction fragments of expected lengths > 10 kb)
- Prepare 1% agarose gel in 0.5x TBE buffer.
- Pour 0.5 TBE buffer to the PFGE unit, precool to 14 °C.
- Set the electrophoresis parameters either using a machine software or manually (e.g., for a DNA fragment range between 10 and 200 kb, appropriate parameters are 6 V/cm, switch time ramped from 1 to 12 sec for 15 h).
- Load the DNA samples in agarose plugs into the gel, load DNA marker.
- After separation, stain the gel with 0.5 μg/ml ethidium bromide in 0.5x TBE for 15 min, document using documentation system. An example of the Nicotiana tabacum DNA separated using PFGE is presented in Figure 1B.
- Transfer DNA to the nylon membrane by capillary or vacuum alkali blotting, hybridize with radioactively labeled telomeric probe (see step C3).
- Prepare 1% agarose gel in 0.5x TBE buffer.
- Conventional agarose electrophoresis (separation of terminal fragments of expected lengths ˂ 10 kb)
- Radioactive labeling of telomeric probe and hybridization
- End-labeling of telomeric oligonucleotide (Note 3)
- Prepare the end-labeling reaction mix at room temperature: 6.5 μl of water, 5 μl of 10 μM synthetic telomeric oligonucleotide, 2 μl of 10×concentrated T4-polynucleotide kinase buffer, 5 μl of 32P-γ-ATP (1.85 MBq), 1 μl of T4-polynucleotide kinase.
- Incubate for 30 min at 37 °C. Heat-inactivate the enzyme (2 min / 94 °C).
- Add the mixture directly to the hybridization solution (see step C3c).
- Prepare the end-labeling reaction mix at room temperature: 6.5 μl of water, 5 μl of 10 μM synthetic telomeric oligonucleotide, 2 μl of 10×concentrated T4-polynucleotide kinase buffer, 5 μl of 32P-γ-ATP (1.85 MBq), 1 μl of T4-polynucleotide kinase.
- Labeling of telomeric concatemers by random priming (Note 3)
- Prepare telomeric concatemers by non-template PCR as described in (Ijdo et al., 1991).
- Perform end-labeling of telomeric concatemers using 32P-α-dATP or 32P-α-dCTP (1.85 MBq) according to instructions in DecaLabel DNA Labeling kit.
- Denature the probe for 10 min at 95 °C, put on ice. Add the probe to pre-heated hybridization solution (see step C3c).
- Prepare telomeric concatemers by non-template PCR as described in (Ijdo et al., 1991).
- Hybridization
- Pre-hybridize the membrane in Hybridization buffer for 1 h at 55 °C in slowly rotating cylinders in hybridization oven. The volume of Hybridization buffer used for pre-hybridization depends on the size of the cylinder; common volume is 30- 50 ml.
- Replace Hybridization buffer by the pre-heated fresh aliquot. Keep the volume of Hybridization buffer as low as possible; common volume ranges between 10-20 ml.
- Add all volume of the radioactively labeled probe (from the C1 or C2) to the Hybridization buffer.
- Hybridize at 55 °C O/N (or at least 16 h).
- Pour out the hybridization solution (to the radioactive waste vessel), add the Washing solution (volume of the Washing solution depends on the size of the cylinder; common volume is 50-80 ml), incubate in the hybridization oven for 15 min at 55 °C. Repeat washing step twice.
- Discard Washing solution, rinse the membrane by 2x SSC and by water and pack the wet membrane to the Saran wrap.
- Expose the membrane to the phosphoimager screen. The time of the exposure depends on the radioactive signal intensity and ranges from 6 h to 24 h.
- Pre-hybridize the membrane in Hybridization buffer for 1 h at 55 °C in slowly rotating cylinders in hybridization oven. The volume of Hybridization buffer used for pre-hybridization depends on the size of the cylinder; common volume is 30- 50 ml.
- End-labeling of telomeric oligonucleotide (Note 3)
- Evaluation of hybridization signals
- Visualize hybridization signals using phosphoimager.
- Evaluate the data using available tools, e.g., TeloTool (Gohring et al., 2014), Telometric (Grant et al., 2001), or TELORUN (Baur et al., 2004). Choosing the appropriate processing tool depends namely on distribution of telomere hybridization signals-see Note 4 for details.
Examples of the TRF analysis are presented in Figure 2A (Arabidopsis thaliana telomeres) and Figure 2B (Nicotiana tabacum telomeres).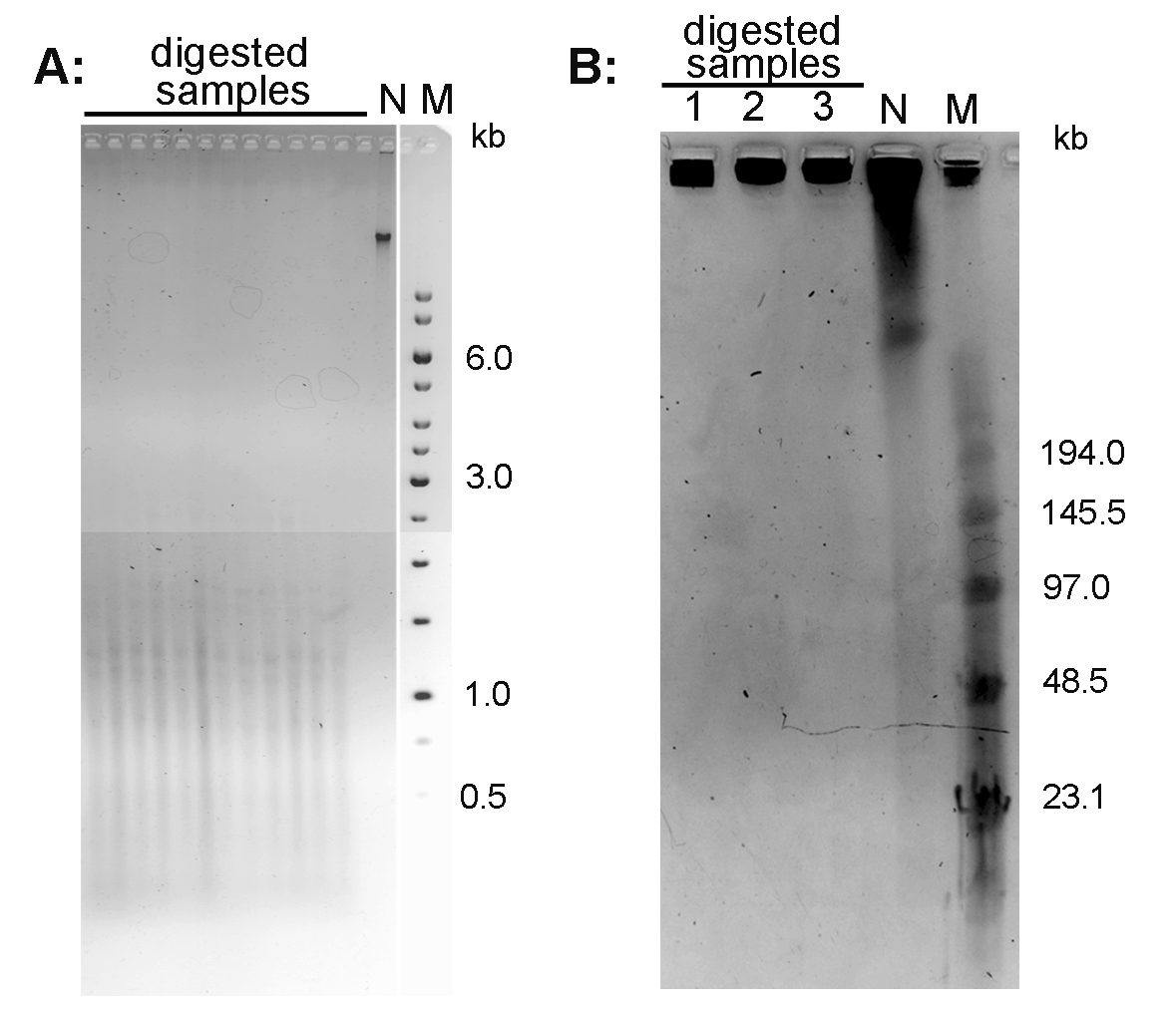
Figure 1. Conventional electrophoresis of Arabidopsis thaliana DNA (A) and pulse field gel electrophoresis of Nicotiana tabacum DNA (B). A. DNA isolated from A. thaliana (ecotype Columbia) leaves was digested with MseI and run on 1% agarose gel in 1x TAE buffer. B. High molecular weight DNA isolated from N. tabacum leaves was digested with TaqI (lane 1), MseI (lane 2) and HinfI + HaeIII (lane 3) and analyzed by PFGE in 0.5x TBE buffer. N-non-digested DNA; M - DNA marker (Low Range PFG Marker).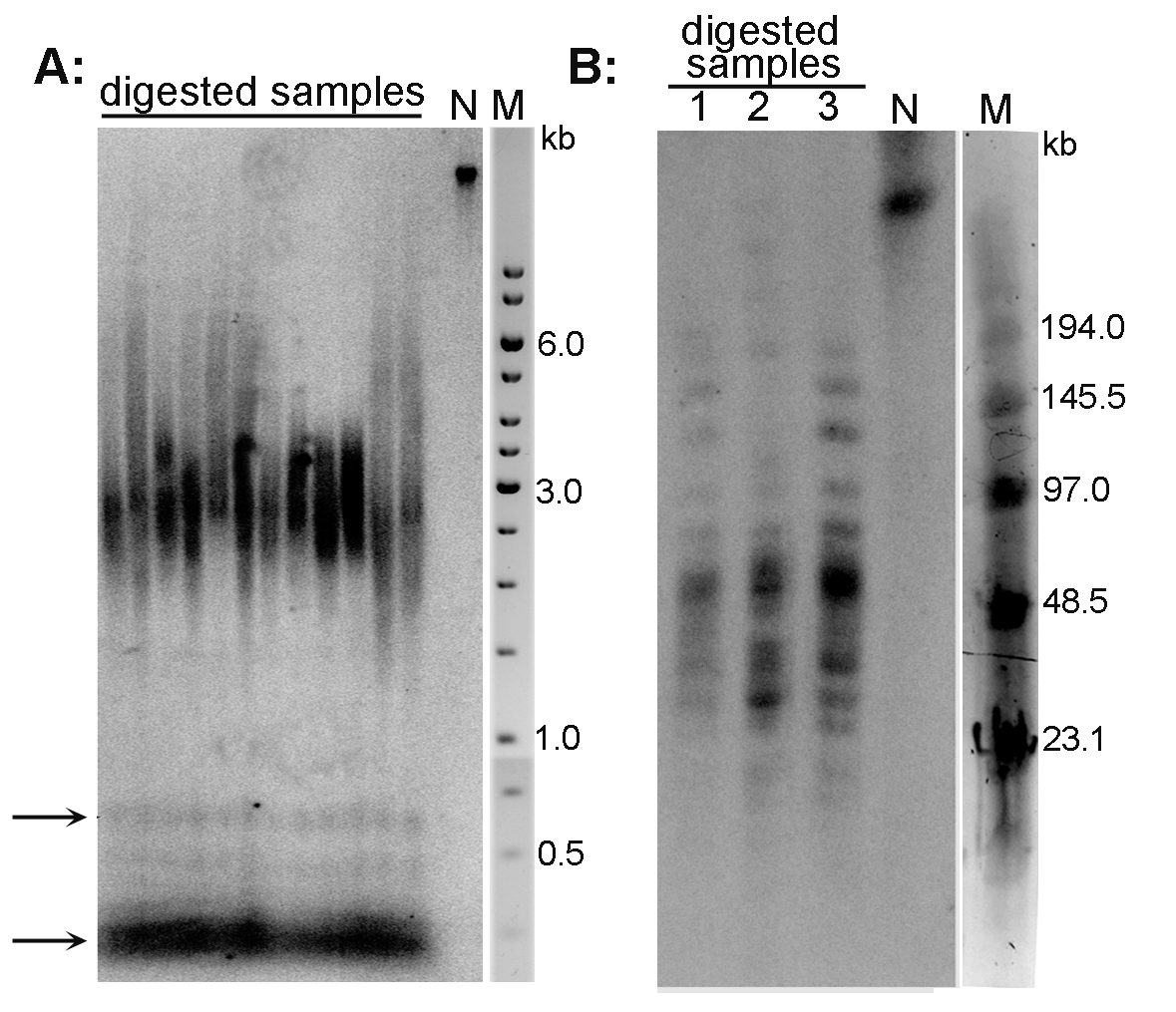
Figure 2. Analysis of telomeres of Arabidopsis thaliana (A) and Nicotiana tabacum (B) plant species by the TRF method. A. DNA isolated from A. thaliana (ecotype Columbia) leaves was digested with MseI, analyzed by conventional agarose electrophoresis and hybridized using radioactively labeled telomeric oligonucleotide. Telomeres of this species are about 3 kb long. Arrows indicate the position of ITR bands; N-non-digested DNA; M-DNA marker (GeneRuler 1 kb DNA Ladder). B. High molecular weight DNA isolated from N. tabacum leaves was digested with TaqI (lane 1), MseI (lane 2) and HinfI + HaeIII (lane 3), analyzed by PFGE and hybridized using radioactively labeled telomeric oligonucleotide. Terminal restriction fragments of tobacco are highly heterogeneous in length (20-160 kb). N-non-digested DNA; M-DNA marker (Low Range PFG Marker).
- Visualize hybridization signals using phosphoimager.
Notes
- To obtain accurate and reproducible data, the integrity of DNA is essential. For analysis of telomere length by conventional electrophoresis (expected telomere length ˂ 10 kb, BAL31 analysis is not performed), use any standard method of DNA isolation (e.g., phenol-chloroform extraction or column-based purification using commercial kits). Check carefully the length of DNA by agarose gel electrophoresis; a sharp band in the compression zone (length > 20 kb) should be visible.
To avoid shearing during isolation, preparation of DNA in agarose plugs is recommended for pulsed-field gel electrophoresis (expected telomere length > 10 kb) and for analysis of the terminal position of telomeric repeats by BAL31 nuclease digestion.
Preparation of high molecular weight DNA in agarose plugs: 1 g of plant tissue is homogenized in liquid nitrogen. Homogenate is mixed with 1 ml of TEM buffer (buffer is preheated in water bath to 45 °C), in the case of thick suspension more TEM buffer is added. Then one volume of 2% low melt agarose in TEM buffer (preheated in a water bath to 45 °C) is added and mixed by pipetting. The mixture is transferred into the disposable plug molds. DNA in agarose plugs is purified by the proteinase K treatment (3x 24 h at 37 °C in the Proteinase buffer, fresh proteinase K is added every 24 h). Proteinase K is inactivated by the PMSF treatment (1 mM PMSF in 1x TE buffer, 10 °C, 3x 30 min) and plugs are washed by 0.1x TE buffer (3x 30 min at room temperature). Before digestion by restriction enzyme or BAL31, high molecular weight DNA in agarose plugs is stored at 4 °C in 0.25 M EDTA. - Be careful and do not use high temperature when handling with DNA embedded in a low melting agarose block. E.g., digestion temperature of commonly used enzyme in TRF, TaqI or Tru1I restriction endonucleases (recognition sequence TCGA and TTAA, respectively), is 65 °C. Incubation of agarose plugs at this temperature leads to their melting and experiment debasement. Temperatures below the optimum (e.g., 55-58 °C) may be used with these enzymes, or such enzymes should be replaced by enzymes with lower temperature optimum (e.g., instead of Tru1I, use its isoschizomer MseI with reaction optimum at 37 °C).
- Two different kinds of probes have been used for probing telomeres in TRF analysis: synthetic telomeric oligonucleotides and concatenated telomeric oligonucleotides (concatemers) (Fajkus et al., 1995; Fajkus et al., 2005; Ijdo et al., 1991; Neplechova et al., 2005; Peska et al., 2015; Sykorova et al., 2003). The advantage of synthetic telomeric oligonucleotides is that the target sequence is well defined. Another advantage is the possibility to use synthetic telomeric oligonucleotides to obtaining strand-specific information, such as the specific detection of overhangs of G-rich telomeric single-stranded DNA (Polanska et al., 2012). The second kind of telomeric probe-concatemers is prepared using PCR and synthetic oligonucleotides designed against both strands of the putative telomere sequence. These are concatenated into a complex probe with extensive heterogeneity in probe length (Ijdo et al., 1991; Peska et al., 2015) but with surprising homogeneity in sequence (Neplechova et al., 2005).
- Telomere lengths (TL) can be evaluated using the following approaches: TeloTool, Telometric 1.2, TELORUN. Each has its own strengths and weaknesses. TeloTool (Gohring et al., 2014) provides very fast, accurate, reproducible and easy-to-use measurements of telomeric signals with automatic detection of sample lanes and markers. After establishing the intensity profiles of sample lanes and markers and probe correction, TL can be computed immediately. This method is based on fitting of Gaussian function to hybridization signal intensity. Fitted Gauss curve is used to compute relevant statistics (mean, standard deviation and range of TL). Hence, TeloTool is suitable for evaluation of TL with normally distributed intensity of hybridization signal. The quality of TL evaluation - how well the curve describes sample data - is described by coefficient of determination. In case of asymmetrically distributed data it is recommended to use Telometric 1.2 (Grant et al., 2001), which allows to compute median of TL, that reflects the real lengths of telomeres more appropriately. On the other hand, Telometric 1.2 may suffer from overestimation of TL. Detailed comparison of both methods is referred in (Gohring et al., 2014). TL can also be measured manually by Multi Gauge signal processing software (FujiFilm) followed by computing statistics. Briefly, using DNA marker as a standard, all sample lanes in hybridization pattern are sectioned into rectangles corresponding to the same molecular weight intervals and unweighted mean telomere length is calculated as Ʃ(ODi × Li)/Ʃ(ODi), where ODi is the hybridization signal intensity (above background) within the interval i, and Li is the molecular weight at the mid-point of the interval i. This method is quite accurate but results may be biased by human factor, namely during rectangle lane sectioning and background subtraction. The computing statistics and its graphical output may be easily generated through TELORUN, an excel sheet with preset computing formulas which is freely available at URL (http://www.swmed.edu/home_pages/cellbio/shaywright/research/sw_lab_methods.htm) (Baur et al., 2004).
Recipes
- BAL31 nuclease buffer
600 mM NaCl
20 mM Tris-HCl (pH 8)
1 mM EDTA
12 mM MgCl2
12 mM CaCl2 - 50x TAE
242 g Tris base
18.61 g EDTA
57.1 ml glacial acetic acid
Water to 1 L - 5x TBE
54 g Tris base, 27.5 g boric acid, 20 ml 0.5 M EDTA (pH 8), water to 1 L - Hybridization buffer
0.25 M NaH2PO4+Na2HPO4 (to get final pH 7.5)
7% SDS, 0.016 M
EDTA - Washing solution
2x SSC [1x SSC: 150 mM NaCl, 15 mM sodium citrate(pH 7)]
0.1% SDS - TE buffer
10 mM Tris-HCl (pH 8)
1 mM EDTA (pH 8) - TEM buffer
1 mM Tris-HCl (pH 8)
50 mM EDTA
0.4 M D-mannitol - Proteinase buffer
0.25 M EDTA (pH 8)
10 mMTris-HCl (pH 8)
1% N-lauroylsarcosine
Acknowledgments
This protocol was adapted from previous work (Fajkus et al., 2008). The work was supported by the Grant Agency of the Czech Republic (13-06595S, 13-06943S) and by the project “CEITEC - Central European Institute of Technology” (CZ.1.05/1.1.00/02.0068) from the European Regional Development Fund.
References
- Baur, J. A., Wright, W. E. and Shay, J. W. (2004). Analysis of mammalian telomere position effect. Methods Mol Biol 287: 121-136.
- Fajkus, J., Dvorackova, M. and Sykorova, E. (2008). Analysis of telomeres and telomerase. In: Hancock, R. (ed). The Nucleus. Humana Press, 267-296.
- Fajkus, J., Kovarik, A., Kralovics, R. and Bezdek, M. (1995). Organization of telomeric and subtelomeric chromatin in the higher plant Nicotiana tabacum. Mol Gen Genet 247(5): 633-638.
- Fajkus, J., Sykorova, E. and Leitch, A. R. (2005). Techniques in plant telomere biology. Biotechniques 38(2): 233-243.
- Gohring, J., Fulcher, N., Jacak, J. and Riha, K. (2014). TeloTool: a new tool for telomere length measurement from terminal restriction fragment analysis with improved probe intensity correction. Nucleic Acids Res 42(3): e21.
- Grant, J. D., Broccoli, D., Muquit, M., Manion, F. J., Tisdall, J. and Ochs, M. F. (2001). Telometric: a tool providing simplified, reproducible measurements of telomeric DNA from constant field agarose gels. Biotechniques 31(6): 1314-1316, 1318.
- Ijdo, J. W., Wells, R. A., Baldini, A. and Reeders, S. T. (1991). Improved telomere detection using a telomere repeat probe (TTAGGG)n generated by PCR. Nucleic Acids Res 19(17): 4780.
- Neplechova, K., Sykorova, E. and Fajkus, J. (2005). Comparison of different kinds of probes used for analysis of variant telomeric sequences. Biophys Chem 117(3): 225-231.
- Peska, V., Fajkus, P., Fojtova, M., Dvorackova, M., Hapala, J., Dvoracek, V., Polanska, P., Leitch, A. R., Sykorova, E. and Fajkus, J. (2015). Characterisation of an unusual telomere motif (TTTTTTAGGG)n in the plant Cestrum elegans (Solanaceae), a species with a large genome. Plant J 82(4): 644-654.
- Polanska, E., Dobsakova, Z., Dvorackova, M., Fajkus, J. and Stros, M. (2012). HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma 121(4): 419-431.
- Richards, E. J. and Ausubel, F. M. (1988). Isolation of a higher eukaryotic telomere from Arabidopsis thaliana. Cell 53(1): 127-136.
- Sykorova, E., Lim, K. Y., Kunicka, Z., Chase, M. W., Bennett, M. D., Fajkus, J. and Leitch, A. R. (2003). Telomere variability in the monocotyledonous plant order Asparagales. Proc Biol Sci 270(1527): 1893-1904.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Fojtová, M., Fajkus, P., Polanská, P. and Fajkus, J. (2015). Terminal Restriction Fragments (TRF) Method to Analyze Telomere Lengths. Bio-protocol 5(23): e1671. DOI: 10.21769/BioProtoc.1671.
Category
Plant Science > Plant molecular biology > Chromatin
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.