- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Infection Experiments (Hepatitis C Virus)




Published: Vol 5, Iss 3, Feb 5, 2015 DOI: 10.21769/BioProtoc.1392 Views: 10053
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The establishment of a cell culture system for hepatitis C virus based on the JFH-1 strain and human hepatoma cell lines has been instrumental for the study of the viral replication cycle. The robustness of the JFH1-based cell culture models enabled many laboratories around the world to perform HCV infections in cell culture, accelerating the identification of cellular and viral targets to develop novel antiviral compounds. Although other robust infection systems based on different molecular clones and different cell lines have been developed since then, here we describe the protocols corresponding to infections with JFH-1 and JFH1-derived viruses carried out in our laboratory to produce virus stocks and persistently infected cell cultures. We also describe the experimental setups used to determine virus spreading capacity (multiple cycle infections) as well as to dissect early and late aspects of HCV infection (single cycle infections).
Materials and Reagents
- Biological materials
- Human hepatoma cells Huh-7 (Nakabayashi et al., 1982) and derived subclone Huh-7.5.1 clone 2, hereafter clone 2 (Pedersen et al., 2007)
- Viruses: Genotype 2a JFH-1 strain (Zhong et al., 2005) and a cell culture-adapted JFH1 variant D183 (Zhong et al., 2006)
- Human monoclonal anti-E2 (AR3A) (antibody provided by Mansun Law, The Scripps Research Institute) (Law et al., 2008)
- Alexa Fluor 555 Goat Anti-Human IgG (H+L) (Alexa 555-conjugated antibody) (Life Technologies, InvitrogenTM, catalog number: A21433 )
- Fetal bovine serum (FBS) (LINUS, catalog number: 501805 )
- BSA (Roche Diagnostics, catalog number: 10 735 086 001 )
- Human hepatoma cells Huh-7 (Nakabayashi et al., 1982) and derived subclone Huh-7.5.1 clone 2, hereafter clone 2 (Pedersen et al., 2007)
- Reagents
- 1 M HEPES (pH 7.4) (Sigma-Aldrich, catalog number: H3375-500G )
- 100x MEM nonessential amino acids (Life Technologies, Gibco®, catalog number: 11140-050 )
- Penicillin/streptomycin (10,000 U/ ml) (Life Technologies, Gibco®, catalog number: 15140-122 )
- 0.5% trypsin/EDTA solution (10x) (Life Technologies, Gibco®, catalog number: 15400-054 )
- Prolong (Life Technologies, catalog number: P-36930 )
- Triton X-100 (Sigma-Aldrich, catalog number: T-8787 )
- Formaldehyde solution (37% wt% in H2O) (Sigma-Aldrich, catalog number: 252549 )
- 4,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, catalog number: 32670 )
- Dulbecco´s Modified Eagle Medium (DMEM) (Life Technologies, Gibco®, catalog number: 41965-039 ) (see Recipes)
- 10x phosphate buffered saline (PBS) (see Recipes)
- Immunofluorescence (IF) buffer (see Recipes)
- 4% formaldehyde solution (see Recipes)
- 1 M HEPES (pH 7.4) (Sigma-Aldrich, catalog number: H3375-500G )
Equipment
- CO2 incubator
- 1.5 ml safe-lock PCR clean microtubes (Eppendorf, catalog number: 0030 123 328 )
- Falcon 50 ml tubes (Falcon®, catalog number: 352098 )
- 75 cm2 cell culture flask (canted neck, 0.2 µM vent cap) (Corning, catalog number: 430641 )
- 162 cm2 traditional straight neck cell culture flask with vent cap (corning, catalog number: 3151 )
- 6 well culture cluster (flat bottom) (Corning, Costar®, catalog number 3506 )
- 12 well culture cluster (flat bottom) (Corning, Costar®, catalog number 3513 )
- 24 well culture cluster (flat bottom) (Corning, Costar®, catalog number 3527 )
- 96 well culture cluster (flat bottom) (Corning, Costar®, catalog number 3599 )
- 5 ml stripette (Corning, Costar®, catalog number 4487 )
- 10 ml stripette (Corning, Costar®, catalog number 4488 )
- 25 ml stripette(Corning, Costar®, catalog number 4489 )
- Pipet aid (IBS INTEGRA Biosciences, catalog number: 155000 )
- Pipetman p200 micropipette (GILSON®, catalog number: F123601 )
- Pipetman p1000 micropipette (GILSON®, catalog number: F123602 )
- Centrifuges (Hettich Zentrifugen, catalog number: EBA 12R )
- -20 °C freezer
- -80 °C freezer
- Fluorescence microscope (inverted fluorescence microscope with long distance objectives)
- Microscope slides and coverslips (Thermo Fisher Scientific, catalog number: 5425508 )
- Filter tips 10 µl (Sorenson Bioscience, catalog number: 35200 )
- Filter tips 20 µl (Sorenson Bioscience, catalog number: 35220 )
- Filter tips 200 µl (Sorenson Bioscience, catalog number: 35230 )
- Filter tips 1,000 µl (Sorenson Bioscience, catalog number: 35260 )
Procedure
Part I. Multiple cycle infections
Multiple cycle infections are also defined as low multiplicity of infection (MOI, i.e. number of infectious particles/cell) experiments. They are generally used to produce virus stocks and to generate persistently infected cell cultures. They also constitute a first approach to characterize the capacity of a virus to spread under different experimental conditions or to compare the spreading capacity of different viruses.
- Virus stock production
Clone 2 cells are used to produce virus stocks because they yield higher infectivity titers in shorter incubation periods, as compared with parental Huh-7 cells.
- Seed 4.5 x 106 Clone 2 cells in 15 ml of complete DMEM in a T162 flask the day before infection.
- Incubate for 24 h in 5% CO2 at 37 °C.
- Dilute the starting virus (JFH-1 or D183) stock in complete DMEM (2 ml/well) to obtain a MOI of 0.01 FFU/cell. [Starting virus stocks are generated by electroporation of in vitro transcribed RNA as described in Zhong et al. (2005).]
- Inoculate by adding the virus dilution (2 ml/well) into the T162 flask.
- Incubate cells in 5% CO2 at 37 °C for 72 h.
- For D183 virus stocks proceed directly to step A13.
- For JFH-1 stocks, wash the cells once with warm PBS and discard.
- Add 1 ml of trypsin/EDTA solution per flask.
- Incubate for 3-5 min at 20-25 °C.
- Add 9 ml of complete DMEM and carefully resuspend the cells.
- Add 50 ml of complete DMEM and split cells into three T162 flasks (20 ml per flask).
- Incubate the cells for additional 72 h in 5% CO2 at 37 °C when producing a JFH-1 stock.
- Collect cell supernatants, centrifuge 1 min at 4,000 rpm at 20-25 °C. Collect supernatants and store at -80 °C in aliquots.
- Replenish cells with complete DMEM (20 ml per T162 flask).
- Incubate for 24 h in 5% CO2 at 37 °C.
- Repeat step A13 through 15 for two-three consecutive days.
- Determine infectivity titer of the samples collected in step A13 (see Part II).
- Seed 4.5 x 106 Clone 2 cells in 15 ml of complete DMEM in a T162 flask the day before infection.
- Generation of persistently infected cell cultures
Persistently HCV infected Huh-7 cells are characterized by constant production of viral RNA, proteins and progeny infectious virus for prolonged periods of time. Although persistently infected cells can be cultured for several months, we recommend limiting the culture passages to less than 8 weeks to reduce the risk for undesired genetic drift of the viral and cell populations (Zhong et al., 2006). Persistent infections can only be established in Huh-7, as more permissive cell lines display severe cytopathology at the peak of infection (Zhong et al., 2006).
- Seed 2 x 106 Huh-7 cells in 10ml of complete DMEM in a T75 flask.
- Incubate for 24 h in 5% CO2 at 37 °C.
- Inoculate cells with JFH-1 at a MOI of 0.01 FFU/cell in a final volume of 10 ml per flask.
- Incubate for a minimum of 12-14 days in 5% CO2 at 37 °C.
- Maintain subconfluent cultures at all times by splitting the cultures every 3-4 days:
- Wash cells once with warm PBS and discard.
- Add 1 ml of trypsin/EDTA solution per flask.
- Incubate for 3-5 min at 20-25 °C.
- Add 5 ml of complete DMEM and resuspend cells in a final volume of 6 ml.
- Discard 4 ml of cell suspension and plate 2 ml into a T75 flask. Add 8 ml of complete DMEM.
- Wash cells once with warm PBS and discard.
- At days 12-14 post-inoculation, the cells will display a marked reduction in their proliferation rate. This occurs at the peak of infection but subsides in the following passages, where persistently infected Huh-7 cells are fully viable but display a slightly longer doubling time as compared with their uninfected counterparts (Zhong et al., 2006).
- Detach the cells from the flask as described in step B5.
- Collect 200 µl of cell suspension and plate it onto a sterile glass coverslip in a M12 plate well. Add 800 µl of complete DMEM and incubate for 24 h. Proceed with this sample to step B10.
- Seed the rest of the cells in a T162 flask. Proceed to step B15.
- Remove supernatant from step 8 and add a 4% formaldehyde solution in PBS (500 µl) onto the glass coverslip.
- Incubate for 20 min at 20-25 °C.
- Wash the cells twice with PBS (1 ml).
- Process the sample to perform immunofluorescence staining (see Part II, step 16).
- Determine the percentage of HCV antigen-positive cells by immunofluorescence microscopy. At day 12-14 post-inoculation nearly 100% of the cells should be HCV antigen-positive.
- Once infection reaches 100% of the cells, the persistently infected cell cultures are maintained subconfluent for a maximum of 8-10 weeks.
In order to maintain subconfluent cultures, split them every 3-4 days as follow:
- Wash cells once with warm PBS and discard.
- Add 1 ml of trypsin/EDTA solution per flask.
- Incubate for 3-5 min at 20-25 °C.
- Add 5 ml of complete DMEM and resuspend cells in a final volume of 6 ml.
- Discard 4 ml of cell suspension and plate 2 ml into a T75 flask. Add 8 ml of complete DMEM.
- Wash cells once with warm PBS and discard.
- Samples of the cells and supernatants are collected according to the experimental design of each experiment (Gastaminza et al., 2008).
- Seed 2 x 106 Huh-7 cells in 10ml of complete DMEM in a T75 flask.
- Determining virus spreading capacity
The following experimental setup enables determining the relative virus spread capacity under different experimental conditions, or to compare the overall growth capacity of different viruses (e.g. mutant viruses). In contrast to single cycle infections (see Part III), this experimental setup does not allow discriminating different aspects of the infection. Thus, determining extracellular progeny virus production is sufficient to characterize the virus spreading capacity. However, we propose collection of additional samples to monitor virus growth using alternative readouts.
- Seed 2 x 105 Huh-7 cells/well in a final volume of 2 ml of complete medium in a 6-well plates one day before inoculation.
- Incubate for 24 h in 5% CO2 at 37 °C.
- Inoculate cells with JFH-1 or D183 virus at a MOI of 0.01 FFU/ cell in a final volume of 2 ml per well.
- Incubate for 2-3 days in 5% CO2 at 37 °C until they reach 90% confluency.
- Collect supernatants and transfer to 1.5 ml tubes. Centrifuge 1 minute at 13,000 rpm. Transfer supernatants to clean 1.5 ml tubes. This sample is used to determine extracellular infectivity (see Part II).
- Wash cells once with warm PBS and discard.
- Add 300 µl of trypsin/EDTA solution per well.
- Incubate for 3-5 min at 20-25 °C.
- Add 600 µl of complete DMEM and resuspend the cells in a final volume of 900 µl.
- Plate 1:3 of the cells (300 µl) in a final volume of 2 ml/well (add 1.7 ml of complete DMEM/well to prepare the next time point of the experiment).
- Transfer 1:3 of the cells (300 µl) to a 1.5 ml tube. Pellet the cells at 3,000 rpm for 5 min at 4 °C. Extract total cellular RNA to determine intracellular HCV RNA levels (Mingorance et al., 2014).
- Transfer 1:3 of the cells (300 µl) to a 1.5 ml tube. Pellet the cells at 3,000 rpm for 10 min at 4 °C. Extract total cellular protein to determine viral antigen levels by western-blot (Mingorance et al., 2014).
- Incubate the cells from step C10 for 3 days in 5% CO2 at 37 °C.
- Repeat as in steps C5-13 until the end of the experiment. Typically sequences of days 2, 4, 6, 8 and 10 or days 3, 6, 9 and 12 should be sufficient to characterize JFH-1 virus spread in Huh-7 cells. In the case of D183, maximum titers are typically reached after day 5-6.
- Seed 2 x 105 Huh-7 cells/well in a final volume of 2 ml of complete medium in a 6-well plates one day before inoculation.
Part II. Virus titration by endpoint dilution and immunofluorescence
This procedure is based on the detection of infection foci (infected cell clusters) by immunofluorescence microscopy. Each infection focus is attributed to one infectious unit, defined as focus forming unit (FFU). The number of infection foci obtained by inoculation of naive cells with serial sample dilutions is used to determine the infectivity titer.
- Seed 104 Clone 2 cells/well in a final volume of 100 µl of complete DMEM in 96-well flat bottom plates. Four to eight wells/sample are required, depending on the expected infectivity titer. Sample replicates are strongly encouraged.
- Incubate for 24 h in 5% CO2 at 37 °C.
- Thaw the infectious samples at 20-25 °C.
- Add 200 µl of complete DMEM to a clean 96-well plate, using 4-8 wells per sample. Set the wells sequentially in a lane to accommodate the different sample dilutions.
- Add 50 µl of the sample onto the first well.
- Discard the tip.
- Homogenize virus-medium dilution with a new filter tip, being careful not to make bubbles.
- Take 50 µl of the mixture and carefully transfer into the next well.
- Repeat steps C5-7 until reaching the desired dilution factor.
- Remove the supernatant of the cells from step C1.
- Add 100 µl of each of the different virus dilutions onto each well.
- Incubate for 72 h in 5% CO2 at 37 °C.
- Discard cell supernatants.
- Add 150 µl of a 4% formaldehyde solution in PBS.
- Incubate for 20 min at 20-25 °C.
- Wash twice with PBS (200 µl).
- Add 30 µl/well of IF buffer.
- Incubate for 1 h at 20-25 °C.
- Prepare a 1 µg/ml dilution of the anti-E2 antibody AR3A in IF buffer.
- Discard IF buffer from the 96-well plate.
- Add 25 µl/well of the anti-E2 antibody dilution.
- Incubate for 1 h at 20-25 °C.
- Prepare a 1:500 dilution of the goat anti-human IgG-Alexa 555 antibody in IF buffer. Add DAPI (1 µg/ml in IF buffer).
- Wash the 96-well plate twice with PBS (200 µl).
- Add 25 µl/well of the secondary antibody dilution prepared as described in step 23.
- Incubate for 1 h at 20-25 °C.
- Wash the 96-well plate twice with PBS (200 µl).
- Add 50 µl of PBS to the test wells. In case of using a glass coverslip, mount onto a microscope slide using ProlongTM.
- Observe under the fluorescence microscope. Count infection foci only in dilutions where they are discrete and well separated from one another, until reaching negative wells (endpoint dilution).
- Calculate the infectivity titer in FFU/ml by, multiplying the number of infection foci found in the different dilutions by the corresponding dilution factor. Multiply by 10 to obtain the foci number per ml (100 µl were used for inoculation). Average the infectivity titers estimated in the different dilutions (typically discrete foci are only observed in 2-3 wells). It is highly recommended to repeat the experiment with different dilutions if the infectivity titer is not calculated out of 2-3 wells or if the endpoint dilution has not been reached.
Part III. Single cycle infection experiments
Single cycle infection experiments, also termed high multiplicity infections (MOI of 10 FFU/cell), attempt modeling synchronic infections of one virion infecting one target cell and are generally used to dissect early and late aspects of the hepatitis C virus life cycle. In contrast to multiple cycle infections, we recommend measuring different parameters such as intracellular and extracellular infectivity and RNA. Simultaneous analysis of these parameters in this experimental setup will enable determining the efficiency of different aspects of the infection. Since high infectivity titer virus stocks are required to perform these experiments and sufficient titers are difficult to reach with the parental JFH-1 strain, we typically use D183 virus stocks for these experiments (see Part I, section A).
- Seed 5 x 104 Huh-7 cells/well in a final volume of 500 µl of complete DMEM in 24-well plates the day before infection. Prepare a different plate per time point (see step 9).
- Incubate for 24 h in 5% CO2 at 37 °C.
- Remove medium and inoculate the cells with D183 virus at MOI of 10 FFU/cell in a final volume of 500 µl per well.
- Keep a sample of the inoculum in order to confirm the multiplicity of infection by infectivity titration (see Part II).
- Incubate for 5 h in 5% CO2 at 37 °C.
- Remove the inoculum and wash cells twice with warm PBS (1 ml).
- Add 500 µl per well of complete DMEM.
- Incubate in 5% CO2 at 37 °C. Typically peak titers are reached around 40 h post-infection. Thus, we recommend collecting samples at 24, 48 and 72 h post-infection. Collecting an additional sample at 5 h, will provide background levels of infectivity and HCV RNA that can be used as a baseline reference for the rest of the time points.
- Collect samples at 5, 24, 48 and 72 h:
- Collect supernatants to determine extracellular infectivity and HCV RNA. Store at -80 °C until analysis (Mingorance et al., 2014).
- Wash cells once with PBS (1 ml) and discard.
- Add trypsin/EDTA solution (100 µl per well).
- Incubate for 3-5 min at 20-25 °C.
- Resuspend cells in 500 µl of complete DMEM per well. Carefully homogenize to prepare a single-cell suspension.
- Transfer 500 µl of the cell suspension to a 1.5 ml tube to determine intracellular infectivity as described in (Gastaminza et al., 2008). Store at -80 °C until analysis.
- Collect 100 µl of the cell suspension in a 1.5 ml tube and process for RNA extraction. Alternatively, store at -80 °C.
- Collect supernatants to determine extracellular infectivity and HCV RNA. Store at -80 °C until analysis (Mingorance et al., 2014).
Representative data
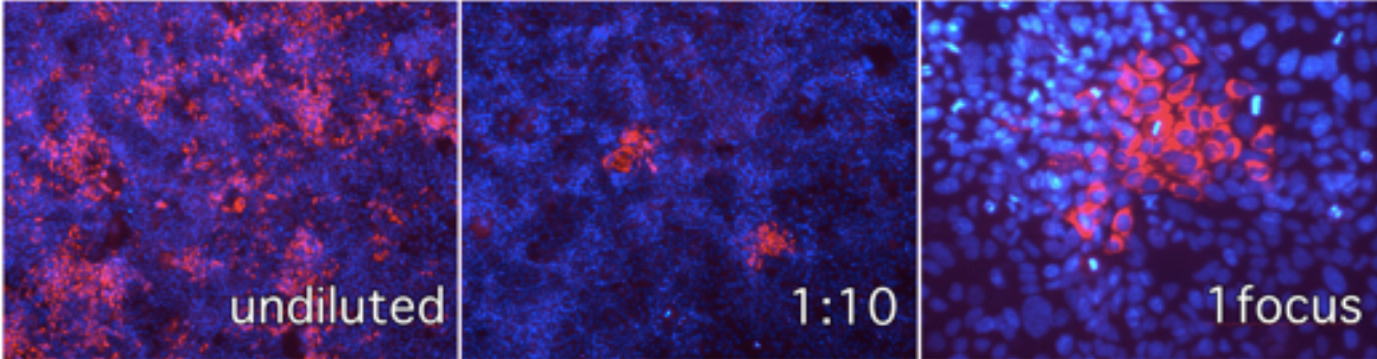
Figure 1. Immunofluorescence microscopy of Huh-7 cells infected with serial dilutions of infected cell supernatant and visualization of individual infection foci in cells inoculated with 1:10 diluted samples. Red channel anti-E2 antibody, blue channel cell nuclei stained with DAPI.
The typical yield of virus stocks should be around 104 FFU/ml for stocks generated with JFH-1 virus and >106 FFU/ml with D183 virus.
Persistently infected cell cultures should produce consistently titers >103 FFU/ml for the first month after reaching 100% infection.
Single cycle infection experiments should display peak extracellular infectivity titers >104 FFU/ml, typically 48 h post-infection.
Notes
- Manipulation of infectious recombinant hepatitis C virus requires specific biosafety conditions and infrastructure. Researchers are advised to revise the degree of biocontainment required by law in the country/region where research will be conducted.
Recipes
- Complete Dulbecco´s Modified Eagle´s Medium (complete DMEM)
DMEM supplemented with 10 mM HEPES
1x non-essential amino acids
100 U/ ml penicillin/streptomycin and 10% fetal bovine serum (heat-inactivated at 56 °C for 30 min)
- 10x phosphate buffered saline (PBS)
Dissolve 80 g NaCl, 2 g KCl, 26.8 g Na2HPO4-H2O and 2.4 g KH2PO4 in 800 ml H2O Adjust to pH 7.4 with HCl
Adjust volume to 1 L with H2O
- Immunofluorescence (IF) buffer
3% BSA
0.3% TritonX-100 in PBS
- 1x trypsin/EDTA (10x) solution
Dissolve 100 ml of 0.5% trypsin/EDTA (10x) solution in 900 ml 1x PBS
- 4% formaldehyde solution
10.8 ml formaldehyde
80 ml H2O and 10 ml 10x PBS
Acknowledgments
This protocol is based on work initially developed at Dr. Francis V. Chisari´s laboratory at The Scripps Research Institute (La Jolla, CA) in collaboration with Dr. Takaji Wakita´s group at the Tokyo Metropolitan Institute of Medical Science (Tokyo, Japan) and was originally generated and improved with the essential contribution of Jin Zhong (currently at Pasteur Institute in Shangai), Sharookh Kapadia (currently at Genentech), Guofeng Cheng (currently at Gilead), Susan Uprichard (currently at U. of Illinois, Chicago) and specially Stefan Wieland (currently at Basel University Hospital Basel).
L.M. was funded by a JAE-Pre fellowship from Consejo Superior de Investigaciones Científicas and CIBERehd (Instituto de Salud Carlos III). C.V. is funded by a Fundación “La Caixa”/CNB fellowship. This work was supported by the grants Plan Nacional De Investigación Científica, Desarrollo e Innovación Tecnológica from the Spanish Ministry of Science and Innovation (SAF2010-19270) and a Marie Curie Career Integration Grant (PCIG-9-GA-2011-293664) from the European Union 7th Framework Programme for Research.
References
- Friesland, M., Mingorance, L., Chung, J., Chisari, F. V. and Gastaminza, P. (2013). Sigma-1 receptor regulates early steps of viral RNA replication at the onset of hepatitis C virus infection. J Virol 87(11): 6377-6390.
- Gastaminza, P., Cheng, G., Wieland, S., Zhong, J., Liao, W. and Chisari, F. V. (2008). Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82(5): 2120-2129.
- Gastaminza, P., Dryden, K. A., Boyd, B., Wood, M. R., Law, M., Yeager, M. and Chisari, F. V. (2010). Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol 84(21): 10999-11009.
- Law, M., Maruyama, T., Lewis, J., Giang, E., Tarr, A. W., Stamataki, Z., Gastaminza, P., Chisari, F. V., Jones, I. M., Fox, R. I., Ball, J. K., McKeating, J. A., Kneteman, N. M. and Burton, D. R. (2008). Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 14(1): 25-27.
- Mingorance, L., Friesland, M., Coto-Llerena, M., Perez-del-Pulgar, S., Boix, L., Lopez-Oliva, J. M., Bruix, J., Forns, X. and Gastaminza, P. (2014). Selective inhibition of hepatitis C virus infection by hydroxyzine and benztropine. Antimicrob Agents Chemother 58(6): 3451-3460.
- Nakabayashi, H., Taketa, K., Miyano, K., Yamane, T. and Sato, J. (1982). Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer research 42(9): 3858-3863.
- Pedersen, I. M., Cheng, G., Wieland, S., Volinia, S., Croce, C. M., Chisari, F. V. and David, M. (2007). Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449(7164): 919-922.
- Zhong, J., Gastaminza, P., Cheng, G., Kapadia, S., Kato, T., Burton, D. R., Wieland, S. F., Uprichard, S. L., Wakita, T. and Chisari, F. V. (2005). Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102(26): 9294-9299.
- Zhong, J., Gastaminza, P., Chung, J., Stamataki, Z., Isogawa, M., Cheng, G., McKeating, J. A. and Chisari, F. V. (2006). Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol 80(22): 11082-11093.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mingorance, L., Vasallo, C., Friesland, M. and Gastaminza, P. (2015). Infection Experiments (Hepatitis C Virus). Bio-protocol 5(3): e1392. DOI: 10.21769/BioProtoc.1392.
Category
Microbiology > Antimicrobial assay > Antiviral assay
Microbiology > Microbe-host interactions > Virus
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.