- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Construction of Glycine Oxidase Mutant Libraries by Random Mutagenesis, Site Directed Mutagenesis and DNA Shuffling

Published: Vol 4, Iss 19, Oct 5, 2014 DOI: 10.21769/BioProtoc.1252 Views: 11021
Reviewed by: Kanika GeraAksiniya AsenovaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Glyphosate, a broad spectrum herbicide widely used in agriculture all over the world, inhibits 5-enolpyruvylshikimate-3-phosphate synthase in the shikimate pathway, and glycine oxidase (GO) has been reported to be able to catalyze the oxidative deamination of various amines and cleave the C-N bond in glyphosate (Pedotti et al., 2009). Here, in an effort to improve the catalytic activity of the glycine oxidase that was cloned from a glyphosate-degrading marine strain of Bacillus cereus (BceGO), we used a bacteriophage T7 lysis-based method for high-throughput screening of oxidase activity and engineered the gene encoding BceGO by directed evolution.
Materials and Reagents
- Bacillus cereus HYC-7
- Escherichia coli (E.coli) DH5α strain, bacteriophage T7
- Glyphosate (Sigma-Aldrich, catalog number: PS1051 )
- Tryptone (Difco)
- Yeast extract (Difco)
- Ampicillin
- o-Dianisidine dihydrochloride (Sigma-Aldrich, catalog number: D3252 )
- Horseradish peroxidase (Sigma-Aldrich, catalog number: P6782 )
- Protein expression vector of pGEX-6P-1 (the plasmid full length 4,984 bp) (GE Healthcare, catalog number: 28-9546-48 ; Genbank accession number: U78872.1)
- Recombinant plasmid pGEX-GO contains encoding gene of glycine oxidase from Bacillus cereus HYC-7
The nucleotide sequence (1,110 bp) was submitted to the NCBI Genbank and gained the accession number (KC203486.1). - Taq DNA polymerase (Takara, catalog number: R500A )
- DpnI restriction enzyme (Takara, catalog number: 1235A )
- dATP, dTTP, dCTP, dGTP (Takara, catalog numbers: 4026Q , 4029Q , 4028Q , 4027Q )
- TransStart® FastPfu DNA polymerase (TransGen Biotech, catalog number: AP221-01 )
- High Pure dNTPs (TransGen Biotech, catalog number: AD101-01 )
- Luria-Bertani medium (see Recipes)
Equipment
- 96 deep-well plates (Axygen, catalog number: P-DW-20-C-S )
- Gel purification column (Axygen)
- Thermo Multiskan spectrum plate reader (Thermo Scientific, catalog number: 51118600 )
- Thermal cyclers (Bio-Rad Laboratories, catalog number: 186-1096 )
- Ultrasonic processor (Sigma-Aldrich, catalog number: Z412619-1EA )
Procedure
- Random mutagenesis
- Prepare the amplification mixture (100 µl) as follows:
10 µl of 10x Taq buffer (Mg2+ plus)
5 µl of 10 mM Mn2+
2 µl of 10 mM dGTP and dCTP
1 µl of 10 mM dATP and dTTP
2 µl of 100 nM oligonucleotide primer F
2 µl of 100 nM oligonucleotide primer R
1 µl of recombinant plasmid pGEX-GO as template
2 µl of Taq DNA polymerase
Add ddH2O to a final volume of 100 μl - The error-prone PCR procedure was performed using the following parameters:
Segment Cycles Temperature Time 1 1 94 °C 3 min 2 30 94 °C 30 sec 59 °C 30 sec 72 °C 80 sec 3 1 72 °C 7 min - Check product by electrophoresis of 5 μl of error-prone PCR product on 1% agarose gel.
- Error-prone PCR products were purified, digested with BamHI and XhoI, cloned into pGEX-6P-1, and transformed into E.coli DH5α to construct the random mutant library.
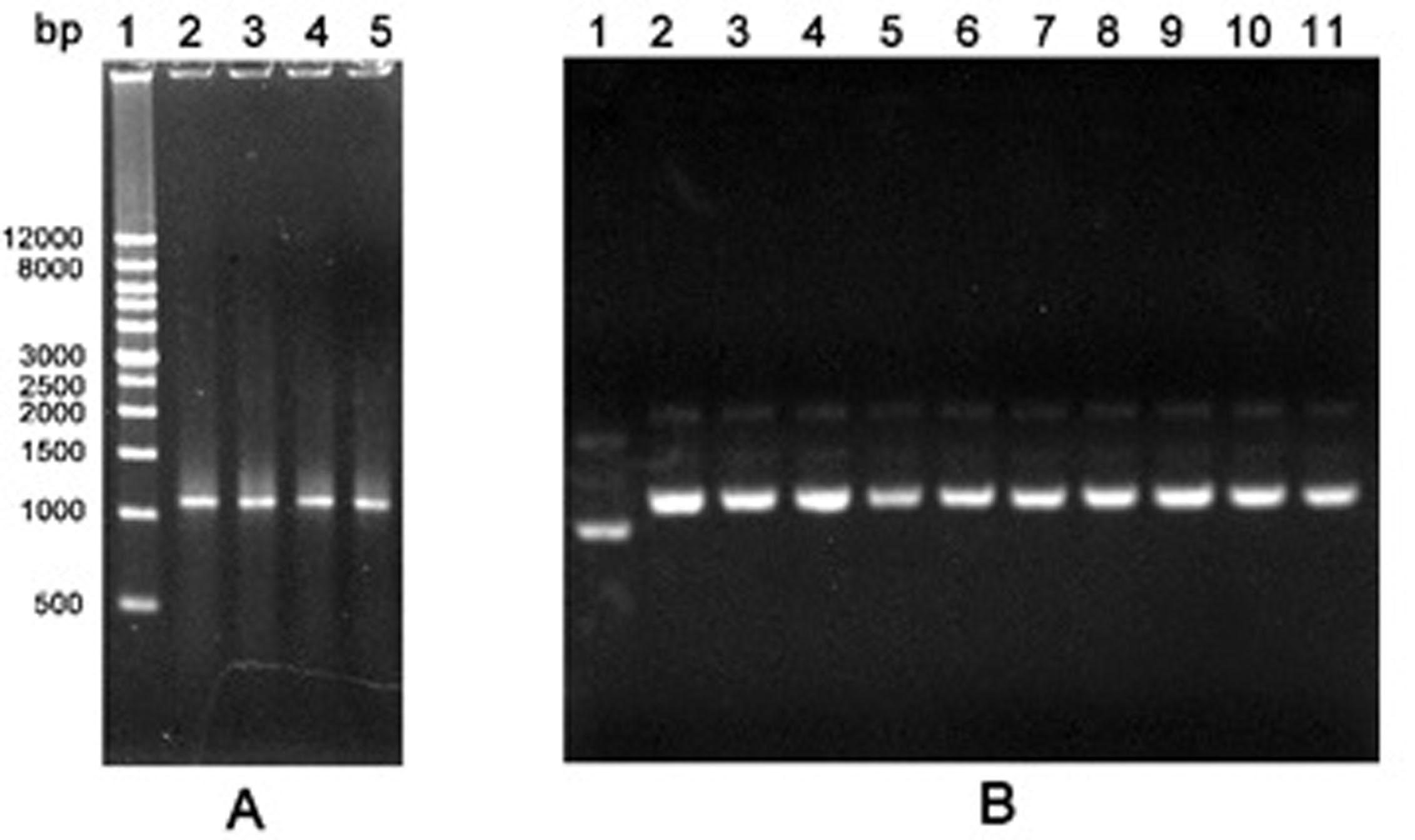
Figure 1. Agarose gel electrophoresis of PCR products by the first round error-prone PCR and recombinant plasmids. A. PCR products. Lane 1: Wide Range DNA Marker (500~12,000 bp); Lane 2-5: Error-prone PCR products; B. Recombinant plasmids. Lane 1: The empty vector pGEX-6P-1; Lane 2-11: Recombinant plasmids pGEX-GOs from colonies.
- Prepare the amplification mixture (100 µl) as follows:
- Site directed mutagenesis
- Prepare the amplification mixture (50 µl) as follows:
10 µl of 10x FastPfu buffer (Mg2+ plus)
1 µl of 10 mM high pure dNTPs
1 µl of 100 nM oligonucleotide primer F
1 µl of 100 nM oligonucleotide primer R
1 µl of dsDNA template
1 µl of FastPfu DNA polymerase
Add ddH2O to a final volume of 50 μl - The site directed mutagenesis PCR procedure was performed as the following parameters:
Segment Cycles Temperature Time 1 1 97 °C 2 min 2 20 94 °C 20 sec 54 °C 30 sec 72 °C 1 min/kb of plasmid length 3 1 72 °C 7 min - Check product by electrophoresis of 5 μl of the site directed mutagenesis PCR product on 1% agarose gel.
- The site directed mutagenesis PCR products were purified, digested with DpnI, and transformed into E.coli DH5α.
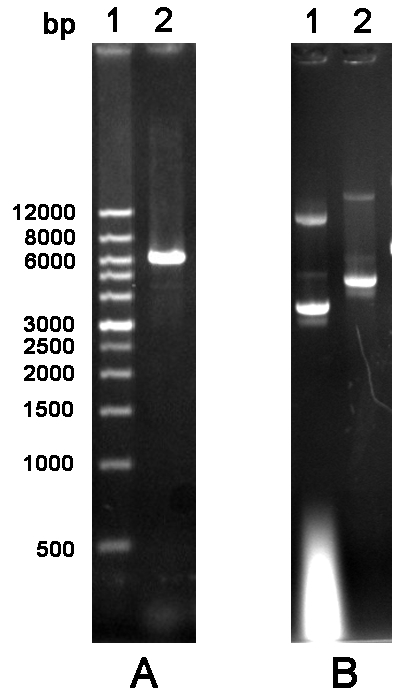
Figure 2 Agarose gel electrophoresis analysis of cylcled PCR product and mutated plasmid. A. PCR product. Lane 1: Wide Range DNA Marker (500~12,000 bp); Lane 2: Cycled PCR product. B. Mutated plasmid. Lane 1: The empty vector pGEX-6P-1; Lane 2: Mutated plasmid.
- Prepare the amplification mixture (50 µl) as follows:
- DNA shuffling
- Obtaining DNA fragments for shuffling.
- Prepare the parental genes by PCR amplification (100 µl) as follows:
10 µl of 10x Taq buffer (Mg2+ plus)
2 µl of 10 mM High Pure dNTPs
2 µl of 100 nM oligonucleotide nested primer F1
2 µl of 100 nM oligonucleotide nested primer R1
1 µl of beneficial mutatant as template
2 µl of Taq DNA polymerase
Add ddH2O to a final volume of 100 μl - The DNA shuffling PCR procedure was performed as the following parameters:
Segment Cycles Temperature Time 1 1 94 °C 3 min 2 30 94 °C 30 sec 62 °C 30 sec 72 °C 80 sec 3 1 72 °C 7 min - Check product by electrophoresis of 5 μl of product on 1% agarose gel.
- Mix ~1 μg of each purified PCR products.
- Prepared for DNA fragmentation by ultrasonic treatment at 0 °C for 40 min to generate a pool of fragments, then the DNA fragments between 100~200 bp were purified using gel purification column.
- Check the DNA fragments by electrophoresis of 10 μl of product on 3% agarose gel.

Figure 3. Schematic of parental genes with the nested primers in the process of DNA shuffling
- Prepare the parental genes by PCR amplification (100 µl) as follows:
- Reassembled by primerless PCR.
- Prepare the amplification mixture (50 µl) as follows:
10 µl of 10x Taq buffer (Mg2+ plus)
2 µl of 10 mM high pure dNTPs
42 µl of purified fragment DNA
1 µl of Taq DNA polymerase
Add ddH2O to a final volume of 50 μl - The primerless PCR procedure was performed as the following parameters:
Segment Cycles Temperature Time 1 1 94 °C 3 min 2 60 94 °C 30 sec 40 °C 30 sec 72 °C 20 sec + 1 sec per cycle 3 1 72 °C 7 min - Check product by electrophoresis of 5 μl of PCR product on 1% agarose gel. A smear of reassembled product that extends above the molecular weight of the parent gene should be visible.
- Prepare the amplification mixture (50 µl) as follows:
- Amplification of full-length sequences.
- Prepare the amplification mixture (50 µl) as follows:
10 µl of 10x Taq buffer (Mg2+ plus)
2 µl of 10 mM high pure dNTPs
1 µl of 100 nM oligonucleotide nested primer F2
1 µl of 100 nM oligonucleotide nested primer R2
5 µl of unpurified reassembly reaction mixture as template
1 µl of Taq DNA polymerase
Add ddH2O to a final volume of 50 μl - The PCR procedure was performed as the following parameters:
Segment Cycles Temperature Time 1 1 94 °C 3 min 2 30 94 °C 30 sec 59 °C 30 sec 72 °C 80 sec 3 1 72 °C 7 min - Check product by electrophoresis of 5 μl of product on 1% agarose gel.
- PCR products were purified, digested with BamHI and XhoI, cloned into pGEX-6P-1, and transformed into E.coli DH5α to create the DNA shuffling library.
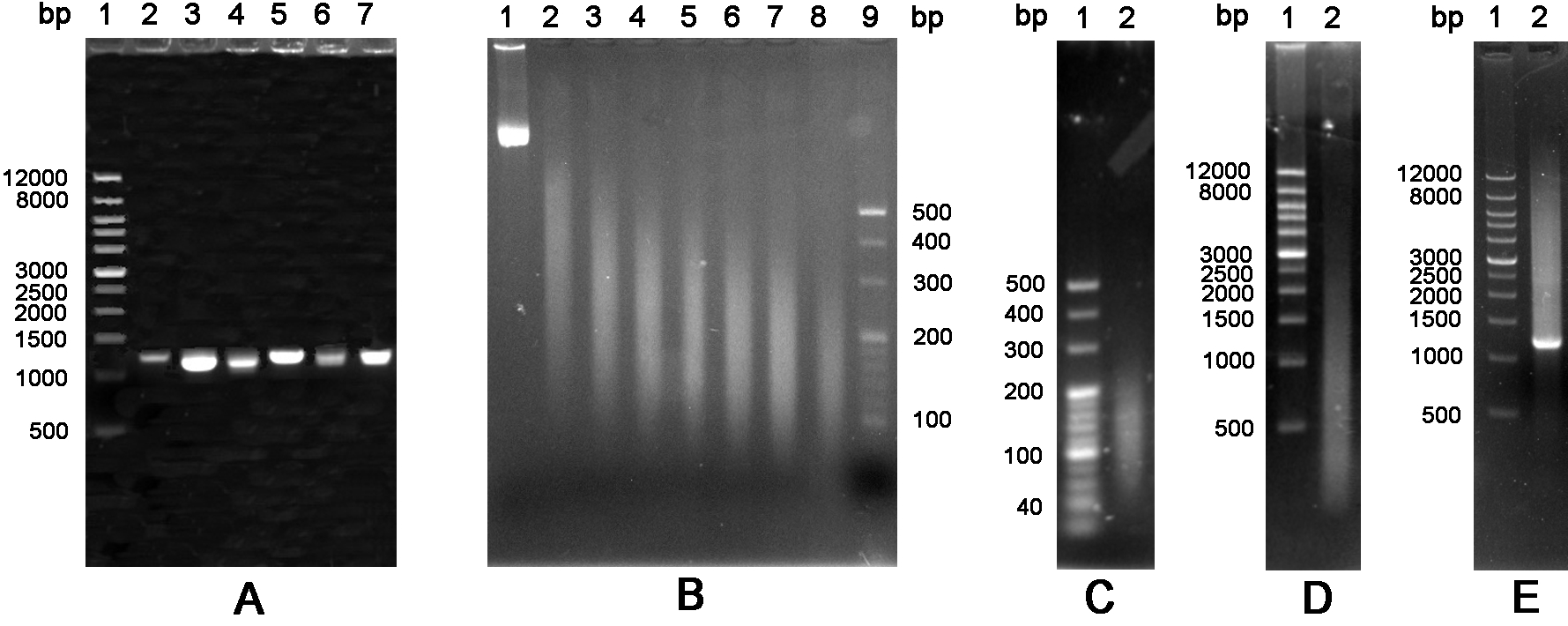
Figure 4. The schematic of glyphosate oxidase gene by DNA shuffling. A. Amplification of six variants GO. Lane 1: Wide Range DNA Marker (500~12,000 bp); Lanes 2-7: DNA fragments encoding variants GO. B. DNA fragmentation. Lanes 1-8: DNA fragments were treated by ultrasonic at 0 °C with a time gradient of 5 min from 0 to 35 min; Lane 9: 20 bp DNA Ladder Marker (20~500 bp). C. Purification of DNA fragments. Lane 1: 20 bp DNA Ladder Marker (20~500 bp); Lane 2: DNA fragments of 100~200 bp were purified from an agarose gel. D. Fragments were reassembled without primers. Lane 1: Wide Range DNA Marker (500~12,000 bp); Lane 2: DNA fragments were reassembled into a full-length gene by 60 cycles without primers. E. The reassembled full-length products were amplified by the standard PCR. Lane 1: Wide Range DNA Marker (500~12,000 bp); Lane 2: PCR product with primers.
- Prepare the amplification mixture (50 µl) as follows:
- Obtaining DNA fragments for shuffling.
- Screening
- The resulting library of BceGO mutants were expressed into 96 deep-well plates (containing 0.6 ml Luria-Bertani medium) and transferred onto Luria-Bertani agar plates as corresponding copies, followed by an overnight growth (37 °C, 300 rpm).
- When the cultures grew to saturation, both IPTG (at a final concentration of 0.1 mM) and the bacteriophage T7 (above 100 particles per cell) were added into 96 deep-well plates to synchronize the induction of recombinant mutants with the release of the lysis of the host E.coli DH5α at 37 °C with shaking for 6 h.
- The enzyme-coupled colorimetric assay (200 µl) was performed as follows:
159 µl of lysis cell extracts
20 µl of 50 mM glyphosate (at a decreasing substrate concentration gradient in sequential rounds of screening system)
20 µl of 0.32 mg/ml o-dianisidine dihydrochloride
1 µl of 5 unit/ml horseradish peroxidase
Then incubated at 25 °C for 8 h - The absorbance change at 450 nm for each well in the microtiter plates was measured and compared with the control (harboring wild-type BceGO or containing the empty vector pGEX-6P-1). Mutants that outperformed the wild-type were chosen for further activity analysis (Figure 1).
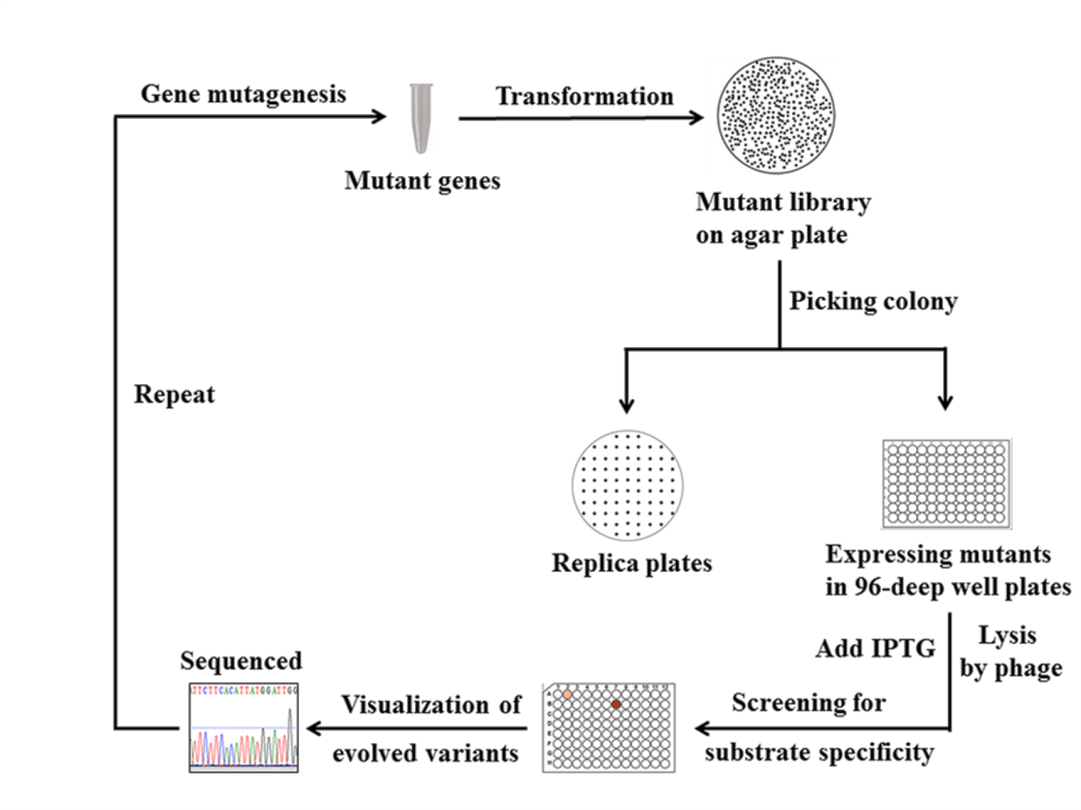
Figure 5. The screening process of glycine oxidase mutant library
- The resulting library of BceGO mutants were expressed into 96 deep-well plates (containing 0.6 ml Luria-Bertani medium) and transferred onto Luria-Bertani agar plates as corresponding copies, followed by an overnight growth (37 °C, 300 rpm).
Representative data
Table 1. The apparent kinetic parameters on glycine and glyphosate measured for wild-type BceGO and variants obtained by random mutagenesis, site saturation mutagenesis and DNA shuffling
| Glycine | Glyphosate | |||
| kcat,app (min-1) | Km,app (mM) | kcat,app (min-1) | Km,app (mM) | |
| Wild-type | 8.17 ± 0.31 | 1.04 ± 0.17 | 5.72 ± 0.42 | 84.79 ± 4.25 |
| 22D11 | 1.16 ± 0.05 | 54.6 ± 3.47 | 2.95 ± 0.21 | 8.29 ± 0.27 |
| 23B1 | 4.56 ± 0.38 | 0.99 ± 0.04 | 7.15 ± 0.62 | 18.88 ± 2.52 |
| B1R | 0.44 ± 0.03 | 58.5 ± 5. 26 | 2.78 ± 0.46 | 2.45 ± 0.15 |
| B2R11 | 1.86 ± 0.09 | 105.6 ± 7.31 | 3.83 ± 0.17 | 2.77 ± 0.21 |
| B2R14 | 1.35 ± 0.12 | 92.5 ± 7. 43 | 4.16 ± 0.14 | 2.17 ± 0.37 |
| B2R23 | 13.02 ± 0.96 | 101.8 ± 8.29 | 30.80 ± 1.33 | 3.80 ± 0.26 |
| B2R81 | 5.41 ± 0.83 | 134.4 ± 10.33 | 7.27 ± 0.75 | 4.37 ± 0.30 |
| B3S1 | 5.43 ± 0.79 | 41.55 ± 3.32 | 11.67 ± 0.98 | 0.53 ± 0.03 |
| B3S4 | 5.68 ± 0.64 | 80.43 ± 5.01 | 12.99 ± 1.14 | 1.37 ± 0.07 |
| B3S6 | 10.14 ± 1.32 | 138.1 ± 12.16 | 13.22 ± 1.78 | 1.69 ± 0.08 |
| B3S7 | 2.30 ± 0.31 | 41.64 ± 2.10 | 4.63 ± 0.39 | 0.57 ± 0.02 |
Especially, B3S1 demonstrated a 160-fold increase in substrate affinity for glyphosate, a 326-fold increase in catalytic efficiency towards glyphosate and a significant enhancement in the specificity constant over the wild-type BceGO, achieving the goal of efficient oxidation of glyphosate by evolution of glycine oxidase.
Notes
- The error-rate is controlled by varying the amount of MnCl2 and unblanced dNTPs added to the error-prone PCR reaction.
- Design nested primer to amplify the parental genes for the DNA fragmentation and full-length sequences from reassembly products.
Recipes
- Luria-Bertani medium
10 g/L tryptone
5 g/L yeast extract
10 g/L NaCl
Ampicillin (50 mg/L) was added as needed.
Acknowledgments
We thank Drs. Ziduo Liu and Dexin Kong for valuable discussions about this article.
References
- GST Gene Fusion System Handbook. (2002). Biosciences, Amersham.
- Zhan, T., Zhang, K., Chen, Y., Lin, Y., Wu, G., Zhang, L., Yao, P., Shao, Z. and Liu, Z. (2013). Improving glyphosate oxidation activity of glycine oxidase from Bacillus cereus by directed evolution. PLoS One 8(11): e79175.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhan, T. (2014). Construction of Glycine Oxidase Mutant Libraries by Random Mutagenesis, Site Directed Mutagenesis and DNA Shuffling. Bio-protocol 4(19): e1252. DOI: 10.21769/BioProtoc.1252.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.