- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Targeted Gene Mutation in Rice Using a CRISPR-Cas9 System


Published: Vol 4, Iss 17, Sep 5, 2014 DOI: 10.21769/BioProtoc.1225 Views: 36472
Reviewed by: Arsalan DaudiFang XuVinay Panwar
Original research article
The authors used this protocol in:

Nov 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
RNA-guided genome editing (RGE) using bacterial type II cluster regularly interspaced short palindromic repeats (CRISPR)–associated nuclease (Cas) has emerged as a simple and versatile tool for genome editing in many organisms including plant and crop species. In RGE based on the Streptococcus pyogenes CRISPR-Cas9 system, the Cas9 nuclease is directed by a short single guide RNA (gRNA or sgRNA) to generate double-strand breaks (DSB) at the specific sites of chromosomal DNA, thereby introducing mutations at the DSB by error-prone non-homologous end joining repairing. Cas9-gRNA recognizes targeted DNA based on complementarity between a gRNA spacer (~ 20 nt long leading sequence of gRNA) and its targeted DNA which precedes a protospacer-adjacent motif (PAM, Figure 1). In this protocol, we describe the general procedures for plant RGE using CRISPR-Cas9 system and Agrobacterium-mediated transformation. The protocol includes gRNA design, Cas9-gRNA plasmid construction and mutation detection (genotyping) for rice RGE and could be adapted for other plant species.
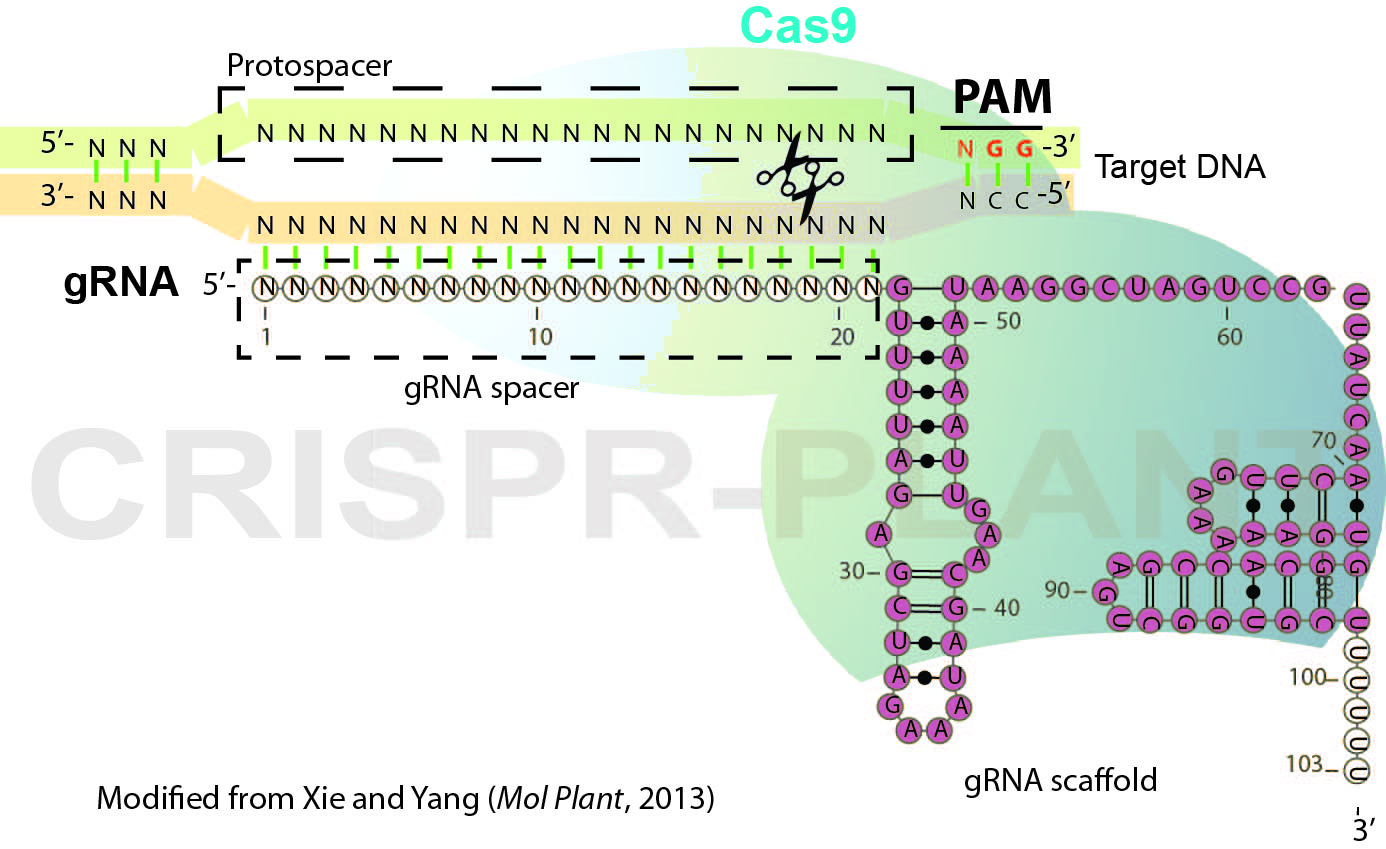
Figure 1. Schematic illustration of CRISPR-Cas9 system
Materials and Reagents
- Oryza sativa L. ssp japonica, Kitaake, Nipponbare, or other cultivars
- Agrobacterium tumefaciens strain EHA105
- pRGEB31 (Xie and Yang, 2013) (Addgene plasmid 51295 , plasmid map is shown in Figure 2)
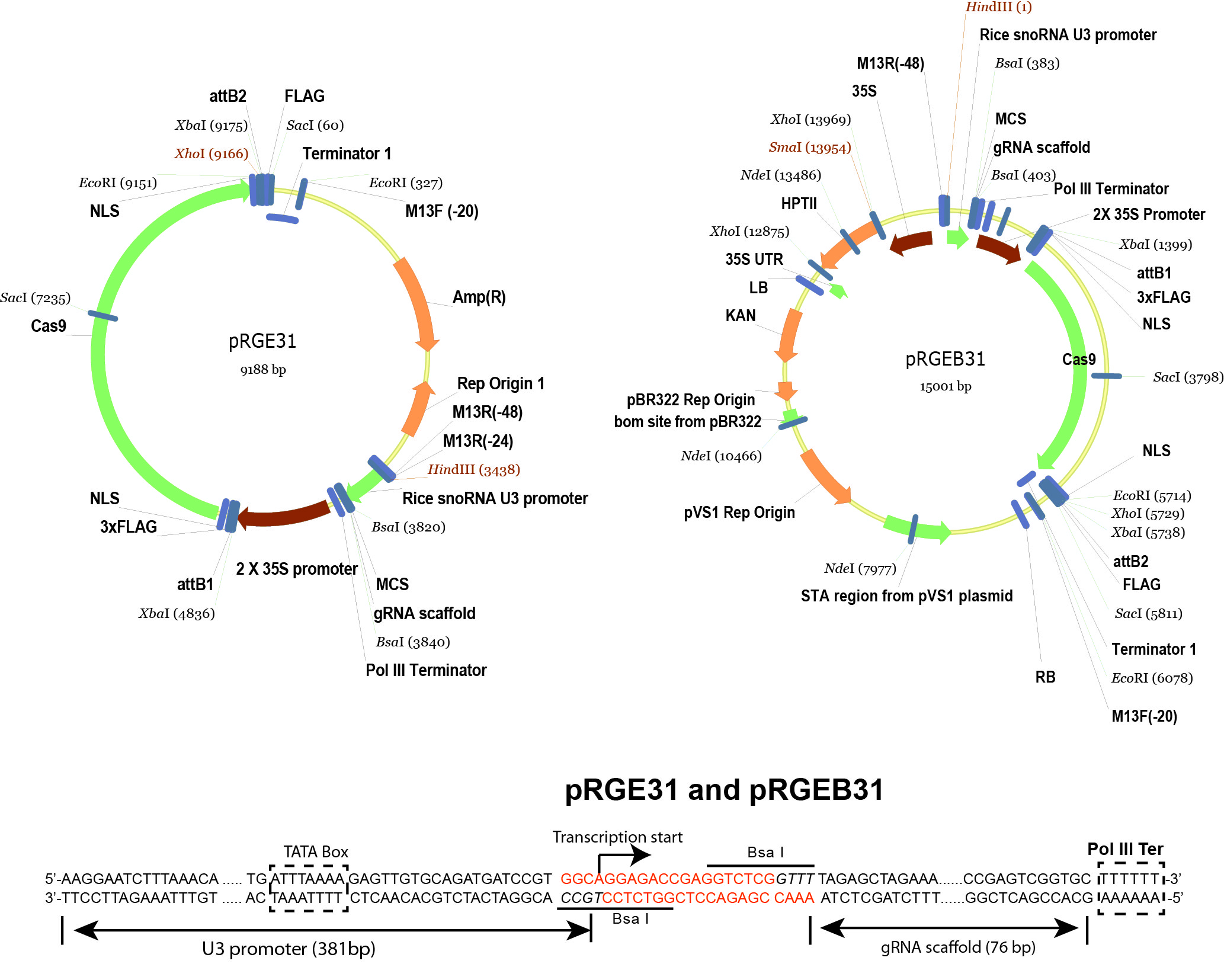
Figure 2. Schematic illustration of pRGE31 and pRGEB31 vectors. The pRGEB31 vector was used in this protocol. - Bsa I (New England Biolabs, catalog number: R0535S )
- 70%, 100% ethanol
- Alkaline phosphatase, calf intestinal (CIP) (New England Biolabs, catalog number: M0290S )
- T4 DNA ligase (New England Biolabs, catalog number: M0202S )
- T4 polynucleotide kinase (T4 PNK) (New England Biolabs, catalog number: M0201S )
- T7 endonuclease I (T7EI) (New England Biolabs, catalog number: M0302S )
- GoTaq DNA polymerase (Promega Corporation, catalog number: M3001 )
- Phusion high-fidelity polymerase (Thermo Fisher Scientific, catalog number: F530S )
- 5x green GoTaq® reaction buffer (Promega Corporation, catalog number: M7911 )
- QIAGEN plasmid mini kit (QIAGEN, catalog number: 12123 )
- QIAquick PCR purification kit (QIAGEN, catalog number: 28104 )
- Hexadecyltrimethylammonium bromide (CTAB) (Sigma-Aldrich, catalog number: H9151 )
- Sodium lauroyl sarcosinate (sarkosyl) (Thermo Fisher Scientific, catalog number: BP235-500 )
- CTAB buffer (see Recipes)
Equipment
- 37 °C water bath
- Thermal cycler
- DNA electrophoresis apparatus
- Microcentrifuge
Procedure
- Design gRNAs to target the genes of interest
- Choose targeted sites and design gRNA spacer sequence as illustrated in Figure 1. For eight model plant species, specific gRNA spacers could be readily designed using the CRISPR-PLANT database (Xie et al., 2014) (http://www.genome.arizona.edu/crispr). Following factors may be considered for designing gRNAs.
- To functionally knock-out the genes of interest, designed gRNAs should introduce DSB close to 5’-end of coding region or located in the essential domains for genes’ function.
- Selected gRNA spacers/target sequence should have sufficient specificity (we recommend that gRNA spacers are ranked with class 0.0 and 1.0 specificity in CRISPR-PLANT database) to avoid off-target editing.
- (Optional) Check gRNA folding, gRNA with three stem-loops as Figure 1 are preferred.
- (Optional) If possible, design a gRNA which would generate mutations at a restriction nuclease enzyme (RE) site in order to introduce RFLP (restriction fragment length polymorphism) for genotyping.
- To functionally knock-out the genes of interest, designed gRNAs should introduce DSB close to 5’-end of coding region or located in the essential domains for genes’ function.
- After selection of targeted sites and spacer sequences, DNA oligos are synthesized to construct the gRNAs as described in Figure 3. Appropriate adaptor sequences should be added for cloning. The reverse primer should contain an adaptor of 5’-AAAC-3’ whereas the forward primer should include an adaptor with 5’-GGCA-3’ (see Figure 3 Case 1, gRNA-spacer starts with A) or 5’-GGC-3’ (Figure 3 Case 2, gRNA-spacer starts with G/C/T).
Note: The gRNAs designed as cases 1 and 2 in Figure 3 showed comparable efficiency in our experiments.
Figure 3. A simple guide for designing DNA oligos to construct the RGE plasmid (Xie et al., 2014)
- Choose targeted sites and design gRNA spacer sequence as illustrated in Figure 1. For eight model plant species, specific gRNA spacers could be readily designed using the CRISPR-PLANT database (Xie et al., 2014) (http://www.genome.arizona.edu/crispr). Following factors may be considered for designing gRNAs.
- Construct gRNA-Cas9 plasmid
- Digest the pRGEB31 vector (see Figure 3 for vector details) by Bsa I.
pRGEB31 2 µg 10x NEB Buffer 4 2 µl 10x BSA 2 µl Bsa I (NEB) 1 µl Add H2O to 20 µl - Incubate at 37 °C for 2-4 h.
- (Optional) Add 0.5 µl of CIP to dephosphorylate the vector by incubating at 37 °C for 30 min.
- Purify the the digested vector using the QIAquick PCR purification kit.
- Prepare DNA oligo-duplex in a PCR tube as following:
Forward oligo (100 uM) 1 µl Reverse oligo (100 uM) 1 µl 10x T4 DNA ligase Buffer 1 µl T4 PNK (NEB) 0.5 µl H2O 6.5 µl
Note: T4 PNK is not required if the vector was not treated with CIP in step 5. - Incubate the tube in a thermal cycler using the following program.
37 °C 60 min 95 °C 10 min Cool down to 25 °C at 0.1 °C/sec - Make a 1: 200 dilution of oligo-duplex for ligation.
- Ligate the diluted oligo-duplex into vector:
Bsa I digested vector n µl (~50 ng) Oligo-duplex (diluted) 1 µl 10x T4 DNA ligase Buffer 0.5 µl T4 ligase (NEB) 1 µl Add H2O to 5 µl - Incubate at room temperature (25 °C) for 2-4 h or at 4 °C overnight.
- Transform E. coli DH5α competent cells (homemade) using 1 µl of ligation product.
- Inoculate 2-4 colonies in LB medium with 50 µg/ml kanamycin.
- Purify plasmids from the transformed DH5α cells using QIAGEN Plasmid Mini kit.
- Confirm pRGEB31-gRNA plasmid constructs by Sanger sequencing using M13R (-48) primer.
- Digest the pRGEB31 vector (see Figure 3 for vector details) by Bsa I.
- Rice transformation
- Introduce the pRGEB31-gRNA plasmid into Agrobacterium strain EHA105.
- Transform rice embryogenic callus using standard Agrobacterium-mediated transformation method
- Introduce the pRGEB31-gRNA plasmid into Agrobacterium strain EHA105.
- Genotyping
Note: The Cas9-gRNA introduced mutation could be examined in hygromycin B selected calli or transgenic plants. To examine Cas9-gRNA introduced mutations, a pair of gene specific primers is required to amplify the targeted region. Because of the high efficiency of Cas9-gRNA system in rice, steps 22 and 23 are optional and the mutation at targeting site could be readily examined by direct Sanger sequencing of PCR product (step 23).- Genomic DNA extraction.
- Ground 10-50 mg of leaves/calli of transgenic rice plants in a 1.5 ml-tube.
- Add 200 µl of heated (60 °C) CTAB buffer to the tube, mixed immediately and incubate at 60 °C for 10 min with occassional shaking.
- Add 80 µl of chloroform, vortex and incubate in an end-to-top rocker at room temperature for 20 min.
- Centrifuge at 14,000 x g for 5 min at room temperature.
- Transfer upper aqueous phase to a fresh tube, add 1/10 Vol of sodium acetate (3 M, pH 5.3) and 2.5 Vol of 100% ethanol, and incubate on ice for 10 min.
- Centrifuge at 14,000 x g for 5 min at room temperature.
- Discard the liquid.
- Add 0.5 ml of 70% ethanol and centrifuge at 14,000 x g for 5min.
- Discard the supernatant liquid and dry the pellet in air.
- Dissolve the genomic DNA in 50-100 µl of H2O.
- Ground 10-50 mg of leaves/calli of transgenic rice plants in a 1.5 ml-tube.
- Setup reactions for PCR amplification using genomic DNAs extracted from transgenic rice and wild type plant.
Rice genomic DNA 100 ng dNTP (10 mM) 1 µl Forward primer (10 μM) 1 µl Reverse primer (10 µM) 1 µl 5x Green GoTaq® Reaction Buffer 10 µl Taq polymerase (2 U/µl) 1 µl Add sterile distilled H2O to 50 µl - Amplify the targeted region by PCR [annealing temperature (x) and extension time (n) depend on primers].
95 °C 3 min 1 cycle 95 °C 15 sec 35 cycles x °C 20 sec 72 °C n min 72 °C 2 min 1 cycle 4 °C Hold 1 cycle - (Optional) If RFLP is introduced (examples was shown in Figure 4)
- Add 5 U of appreciate restriction enzyme to 25 µl of PCR product (~200 ng).
- Incubate at 37 °C for 2-4 h.
- Analyze the digested DNA fragment by 1% agarose gel electrophoresis.
- Add 5 U of appreciate restriction enzyme to 25 µl of PCR product (~200 ng).
- (Optional) If no RFLP was introduced, use T7E1 assay to detect mutations:
Note: High fidelity DNA polymerase (Phusion DNA polymerase) should be used in step 19 for T7E1 assay. T7E1 assay could not detect homozygous mutation. Examples (line #1 and #3) were shown in Figure 4.- Purify the PCR products using PCR purification column.
- Set up the T7E1 assay in PCR tubes as following:
PCR product 100-200 ng 10x NEB buffer 2 1 µl Add H2O to 9.5 µl - Denature and anneal PCR products using the following program:
95 °C 10 min Ramp to 85 °C at 0.1 °C/sec - 85 °C 5 min Ramp to 65 °C at 0.1 °C/sec 65 °C 2 min Ramp to 45 °C at 0.1 °C/sec 45 °C 2 min Ramp to 25 °C at 0.1 °C/sec 25 °C hold - Add 0.5 µl of T7E1 to the annealed PCR product.
- Incubate the reaction at 37 °C for 1 h.
- Analyze the digestion by agarose gel electrophoresis.
- Purify the PCR products using PCR purification column.
- Clone and sequence the PCR product (at least 4 colonies) to confirm the mutation in transgenic plants.
Note: T7E1 enzyme is not absolutely mismatch specific; it also nicks dsDNA slowly. Therefore, it is important to control the enzyme amount and reaction time and carefully interpret the results in T7EI assay.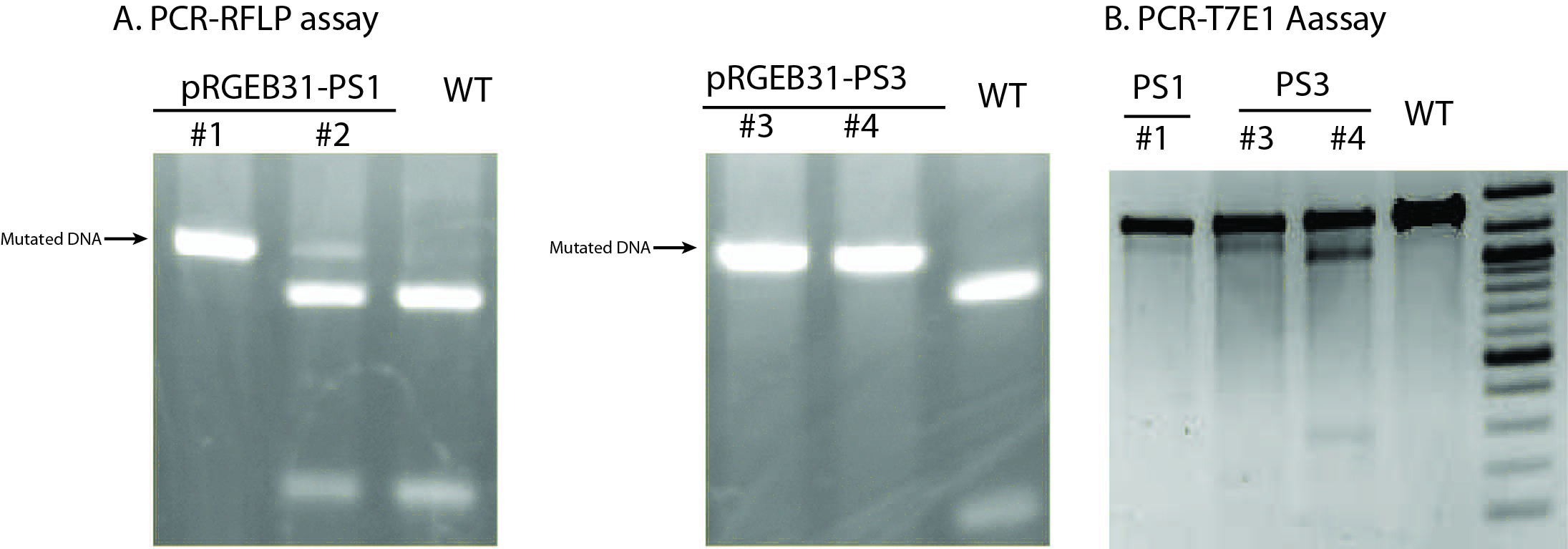
Figure 4. Examples of PCR-RFLP assay (A) or T7E1 assay (B) using T0 transgenic plants. In PCR-RFLP assay, mutated DNA is resistant to RE digestion whereas wild type DNA would be cut into pieces. No wild type DNA was detected in transgenic lines #1, #3, and #4 (T0 generation) based on PCR-RFLP results, suggesting highly efficient mutation rate of RGE in rice. Of note, the size of extra band from line #1 and #3 in the PCR-T7E1 assay was different from the expected size in line #4, which was likely resulting from imperfect specificity of T7E1 enzyme. Based on these assays and final sanger sequencing results, lines #1 and #3 were found to contain homozygous, identical mutations at the targeted site whereas line #4 contained heterozygous, different mutations at the targeted site.
- Genomic DNA extraction.
Recipes
- CTAB buffer (100 ml)
Dissolve2.56 g of sorbitol, 1 g of Sarkosyl and 0.8 g of CTAB in 60 ml H2O.
Add 11 ml of Tris buffer (pH 8.0, 1 M), 4.4 ml of EDTA (pH 8.0) and 16 ml of NaCl (5 M)
Heat the solution briefly to dissolve CTAB and Sarkosyl
Add dH2O to 100 ml
Autoclave and stored at room temperature
Acknowledgments
This work was supported by Pennsylvania State University and a research grant from NSF Plant Genome Research Program (DBI-0922747) to YY.
References
- Xie, K. and Yang, Y. (2013). RNA-guided genome editing in plants using a CRISPR-Cas system. Mol Plant 6(6): 1975-1983.
- Xie, K., Zhang, J. and Yang, Y. (2014). Genome-wide prediction of highly specific guide RNA spacers for the CRISPR-Cas9 mediated genome editing in model plants and major crops. Mol Plant 7(5): 923-926.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Xie, K., Minkenberg, B. and Yang, Y. (2014). Targeted Gene Mutation in Rice Using a CRISPR-Cas9 System. Bio-protocol 4(17): e1225. DOI: 10.21769/BioProtoc.1225.
Category
Plant Science > Plant molecular biology > RNA > RNA interference
Molecular Biology > RNA > mRNA translation
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.