- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Ciliary and Flagellar Membrane Vesicle (Ectosome) Purification
Published: Vol 4, Iss 12, Jun 20, 2014 DOI: 10.21769/BioProtoc.1156 Views: 10793
Reviewed by: Ru ZhangLin FangFanglian He
Original research article
The authors used this protocol in:

Jan 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Eukaryotic cilia/flagella are ideal organelles for the analysis of membrane trafficking, membrane assembly, and the functions of a variety of signal transduction molecules. Cilia are peninsular organelles and the membrane lipids, membrane proteins, and microtubular-associated components are selectively transported into cilia through the region formed by the basal body/transition region and tightly associated ciliary membrane. Cilia can be isolated from many organisms without disrupting cells and many will rapidly regenerate cilia (with the ciliary membrane lipids and proteins) to replace those that are released. Despite their ease of isolation, we have relatively little understanding of the mechanisms that regulate lipid and protein transport into ciliary membranes (Pazour and Bloodgood, 2008; Bloodgood, 2009; Bloodgood, 2012).
Chlamydomonas flagella shed membrane vesicles, also called ectosomes (Wood et al., 2013) from flagellar tips and these vesicles can be purified from the culture medium without damaging or deflagellating cells (McLean et al., 1974; Bergman et al., 1975; Snell, 1976; Kalshoven et al., 1990). Based on a comparison of biotinylated proteins on the shed vesicles with biotinylated proteins isolated from purified flagella and cell bodies, the ectosomes contain most, but not all, flagellar surface proteins and none of the major cell body proteins (Dentler, 2013). Although ectosomes have only been purified from Chlamydomonas cells, preliminary evidence indicates that similar vesicles are released from Tetrahymena cilia (Dentler, unpublished).
Flagellar (and ciliary) membranes or membrane proteins also can be released from purified flagella/cilia. Most membrane proteins can be solubilized by extracting purified cilia with nonionic detergent [Triton X-100 or X-114 or Nonidet P-40 (NP-40)] and pelleting the microtubules (axonemes). However, not all membranes are released by detergent (Dentler, 1980) and the supernatant also contains all of the flagellar proteins that are not attached to the microtubules.
Intact membrane vesicles can be released from flagella by agitation of flagella, often with low concentrations of nonionic detergents or freeze-thawing (Witman et al., 1972; Snell, 1976; Dentler, 1980; Dentler, 1995; Bloodgood and May, 1982; Pasquale and Goodenough, 1987; Iomini et al., 2006; Huang et al., 2007). Once released, they can be purified from axonemes by differential centrifugation.
Each of these methods may enrich for different populations of axonemal and membrane proteins and lipids. The different solubility of membranes may reveal local differences in lipid or protein composition (Bloodgood, 2009). The ectosomes contain most but not all surface proteins found on purified Chlamydomonas flagella (Dentler, 2013). The ectosomes vesicles may be enriched in different soluble flagellar proteins than those trapped as vesicles are released from purified flagella. The detergent-solubilized “membrane+matrix” will contain all soluble membrane proteins as well as all of the soluble proteins in the flagellar compartment.
In this paper, a method to purify ectosomes vesicles released from the tips of living Chlamydomonas cells is presented as are two methods to release flagellar membrane vesicles and proteins from purified flagella.
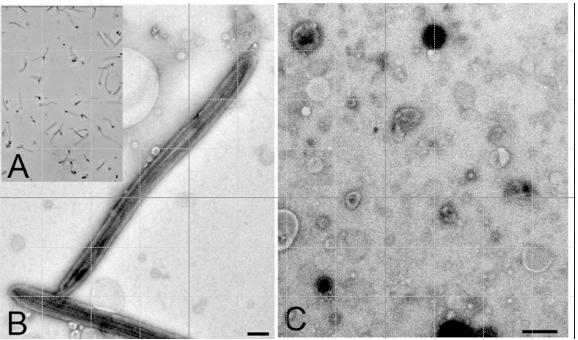
Figure 1. A: Purified flagella (phase contrast); B: Purified flagella (TEM); C: Shed membrane vesicles (TEM). Bars = 500 nm
Materials and Reagents
- Cells
Chlamydomonas cells can be obtained from a variety of sources and pure strains can be obtained from the Chlamydomonas Resource Center (http://chlamycollection.org/contact-us/). - Media and cell culture (see Notes)
- Culture media for washing cells (2-4 L)
- 0.5 N acetic acid
- 0.5 N KOH
- Sucrose
- Triton X-100 or Nonidet P-40
- Cilia wash buffer (CWB) (see Recipes)
Equipment
- Pellicon tangential flow microfiltration cassettes (Millipore, www.millipore.com)
Note: Cells can be harvested using 450 ml centrifuge bottles and large rotor but, for 8-16 L of cells, harvesting is more rapid and fewer cells are deflagellated using a Pelicon. - Preparative centrifuge with rotors for 450 ml bottles and for 40 ml tubes
- 450 ml centrifuge bottles
- 40 ml centrifuge tubes
- 12 ml centrifuge tubes
- Medium speed centrifuge with swinging bucket rotor and angle rotor
- Fernbach flasks (2,800 ml) or large flasks
- Ultracentrifuges and rotors
Note: For 750 ml, I use Beckman 35 or Ty45Ti rotors and two Beckman ultracentrifuges. - Phase contrast microscope
- Orbital shaker
- Bright fluorescent light - generally 4-6 F20/40 PL/AQ lamps
- Transmission electron microscope
- Magnetic stirrer
- pH electrode
Procedure
Method 1. Purification of ectosomes
Ectosomes are released by flagella during the incubation period and contain most of the surface-exposed (biotinylated) proteins found on purified flagella (Dentler, 2013).
- Harvest 8-16 liters of cells using a Pellicon cassette system. Further concentrate cells by centrifugation (1,500 x g, 4 min) if necessary.
- Gently suspend the cell pellets in fresh culture medium.
- To avoid deflagellating cells, suspend pellets by gentle swirling or by agitation with a large bore glass or plastic pipette. The total suspension volume depends on the availability of centrifuges.
- Each Beckman 35 or 45Ti rotor can hold six 70 ml tubes, for a total volume of 490 ml/centrifuge, so suspend cells in a total volume of 700-900 ml of M medium. If there is access to larger capacity centrifuges or additional centrifuges and rotors, it is best to increase the volume of cells to avoid crowing.
- Examine cells by phase contrast microscopy to insure that there are no dividing cells and that nearly 100% of the cells are fully flagellated and motile. If cells are sick or not fully flagellated, the preparations will be contaminated by cell debris or released flagella and it’s best not to proceed with these cells.
- To avoid deflagellating cells, suspend pellets by gentle swirling or by agitation with a large bore glass or plastic pipette. The total suspension volume depends on the availability of centrifuges.
- Incubate suspended cells in constant light for 6 h or longer.
- Cells can be aerated with a sintered glass bubbler in 1 L bottles or can be gently swirled in 2,700 ml Fernbach flasks using an orbital shaker under bright fluorescent light.
- At the end of the incubation period, check cells by microscopy to insure that all are intact, flagellated, and motile. At the end of the incubation period, pellet the cells by centrifugation for 1,500 x g, 4 min. This can be done at 4 °C or at room temperature (~23 °C), the temperature at which the cells were incubated.
- Carefully decant the supernatant (containing shed vesicles) to avoid disturbing the cell pellet or aspirate the supernatant from the bottles.
- Examine the supernatant by phase contrast microscopy to insure that no flagella are present. If flagella or cells are present, centrifuge the supernatant at 11,000 x g for 10 min. The cell pellets can be gently suspended in M medium and deflagellated to purify flagella (see below).
- Carefully decant the supernatant (containing shed vesicles) to avoid disturbing the cell pellet or aspirate the supernatant from the bottles.
- Pellet the shed membranes by centrifuging the supernatant for 60 min, 125,000 x g, 4 °C.
- Remove the supernatant by aspiration. The vesicle-containing pellets are nearly transparent and are easily dislodged. Often, there is a tight green pellet containing cell bodies or chloroplasts that is overlain by a more transparent pellet. Try to suspend the membrane-containing transparent material without disturbing the green pellet. Suspend in a small volume of CWB, keeping the combined volume of all suspended pellets less than 2 ml.
- Layer the suspended pellets over ~ 8 ml of CWB with 25% sucrose and centrifuge 3,000 x g, 10 min, 4 °C, in a swinging bucket rotor.
- Remove the layer (containing vesicles) above the 25% sucrose/CWB and centrifuge at 46,000 x g for 30 min, 4 °C to pellet membrane vesicles. Suspend the pellet in a small volume of CWB and check for flagella or cell bodies by phase contrast and by negatively staining and examining by transmission electron microscopy.
Method 2. Purification of vesicles from isolated flagella
- Flagellar purification - pH shock
Flagella can be isolated using dibucaine or pH shock but I’ve found the membrane to be more intact in pH shocked flagella. For other methods to isolate and fractionate cilia and flagella from various organisms, see Methods in Cell Biology, Vol 47, 1995.- Harvest cells using a pelicon harvester and by centrifugation (see above) and suspend cells in fresh M medium to a final volume of 100-200 ml. The cells used to isolate released membrane vesicles (above) also can be used to isolate flagella.
- Using a magnetic stirrer and calibrated pH electrode, deflagellate cells by lowering the pH to 4.0 by dropwise addition of 0.5 N acetic acid. Do not leave cells at pH 4 for longer than 1-1.5 min. Raise the pH to 7.0 by dropwise addition of 0.5 N KOH. For 100 ml of cells in M medium, use approximately 4.5 ml of 0.5 N acetic acid and 5.2 ml of 0.5 M KOH. For most purposes, it is best to monitor the pH change but, for mass-deflagellations in microtitre plates, adding defined quantities of acetic acid and KOH is useful.
- Pellet cells by centrifugation at 1,100 x g, 3 min.
- Aspirate the supernatant (containing flagella) with a large bore pipette and layer over CWB with 25% sucrose.
- Centrifuge in a swinging bucket rotor at 2,500 x g for 10 min, 4 °C.
- Flagella will collect at the interface above the 25% sucrose.
- Centrifuge in a swinging bucket rotor at 2,500 x g for 10 min, 4 °C.
- Aspirate the white flagella layer, dilute with CWB, and pellet flagella at 17,000 x g, 20 min, 4 °C.
- Suspend flagella in CWB using a large bore pipette to avoid breaking flagella and check with phase contrast microscopy to insure the preparation is free of cell bodies or other debris.
- Harvest cells using a pelicon harvester and by centrifugation (see above) and suspend cells in fresh M medium to a final volume of 100-200 ml. The cells used to isolate released membrane vesicles (above) also can be used to isolate flagella.
- For detergent-solubilized membrane + matrix.
Detergent-extraction of cilia and flagella solubilizes most, but not all, of the membrane but also releases soluble ciliary proteins. Bonafide flagellar trans-membrane proteins can be unambiguously identified if they are biotinylated on intact cells (see Reference 7).- Suspend the flagella in CWB, add 10% Triton X-100 or Nonidet P-40 to a final concentration of 1%, incubate 10-20 min, 4 °C.
- Centrifuge for 15 min at 27,000 x g, 4 °C to pellet the detergent-insoluble axonemes and detergent-insoluble membranes. The solubilized membrane and matrix proteins will be in the supernatant.
- Suspend the flagella in CWB, add 10% Triton X-100 or Nonidet P-40 to a final concentration of 1%, incubate 10-20 min, 4 °C.
- Vesicle release and purification
Purification of membrane vesicles should insure that all of the proteins and lipids are components of the membrane but not the soluble flagellar matrix proteins. However, contamination of the vesicles with soluble flagellar proteins is possible as the membranes are released from flagella and vesiculate, trapping flagellar matrix proteins. Additionally, rubbing membranes off axonemes or freeze-thawing them leads to considerable breakage of the axonemal microtubules and release of axonemal components. Detergent extraction can be done with no significant axonemal breakage.
This is a modification of the method used to purify Tetrahymena ciliary membranes (Dentler, 1980; Dentler, 1995). Other methods to release membranes from purified flagella include freeze-thawing and rubbing off membranes (Wang and Snell, 2003; Huang et al., 2007; Pasquale and Goodenough, 1987; Iomini et al., 2006; Snell, 1976) or treatment with low concentrations of detergents (Witman et al., 1972; Bloodgood and May, 1982).- Start with purified flagella (above) suspended in CWB.
- Agitate the flagella by shearing with a glass pipette or by vortexing for 1 min, incubate on ice 1 min, and vortex again. Repeat 5-10 times.
- Examine by phase contrast microscopy to assay for the formation of small membrane vesicles (will appear as dots) and the membrane-free axonemes (which will be thin and less refractile than intact cilia).
- If membranes cannot easily be released, add Nonidet P-40 or Triton X-100 to a final concentration of 0.01-0.02% during vortexing.
- Layer the sheared or vortexed flagella over sucrose step gradients composed of 1 ml 20% sucrose (w/v) in CWB, 1 ml of 30% sucrose (w/v) in CWB, and 1.2 ml of 40% sucrose (w/v) in CWB.
- Centrifuge in a swinging bucket rotor. Centrifuge samples at 200,000 x g, 2 h and collect membranes from the interface between the 30% and 40% sucrose layers (Dentler, 1995).
Note: Alternatively, vesicles may be separated from axonemes by layering the sheared flagella over ~8 ml of CWB with 25% sucrose and centrifuging 3,000 x g, 10 min, 4 °C as was done for the released membranes (above). The axonemes with attached membranes will be in the pellets. - Carefully remove membranes from the interfaces, dilute with cold CWB, and pellet vesicles by centrifugation at 48,000 x g for 1 h.
- Suspend the vesicles in a small amount of CWB and examine by phase contrast and/or negative staining and transmission electron microscopy to insure that the vesicles are intact and are not contaminated with cell debris or axonemes.
- Start with purified flagella (above) suspended in CWB.
Notes
- Notes about media and cell culture
- Culture media recipes are available at http://www.chlamy.org/media.html. I prefer to culture cells in Sager and Granick minimal (M) medium (http://www.chlamy.org/SG.html) but TAP medium is widely used (http://www.chlamy.org/TAP.html). Media preparation and culture conditions also are described in References 8 and 15.
- Autoclave media in 4 L Pyrex bottles. Aerate cells with a sintered glass filter and house air. Culture in constant light or, to obtain a uniform population of synchronized cells, culture in M medium on a 12 h light/dark cycle.
- For most preparations, I use 8-16 liters of cells grown to ~ 5 x 105 cells/ml.
- Culture media recipes are available at http://www.chlamy.org/media.html. I prefer to culture cells in Sager and Granick minimal (M) medium (http://www.chlamy.org/SG.html) but TAP medium is widely used (http://www.chlamy.org/TAP.html). Media preparation and culture conditions also are described in References 8 and 15.
Recipes
- Cilia wash buffer (CWB)
50 mM Tris-HCl (pH 7.4)
3 mM MgSO4
100 mM EGTA (pH 7.4)
250 mM sucrose
Acknowledgments
This protocol was adapted from Dentler (2013).
References
- Bergman, K., Goodenough, U. W., Goodenough, D. A., Jawitz, J. and Martin, H. (1975). Gametic differentiation in Chlamydomonas reinhardtii. II. Flagellar membranes and the agglutination reaction. J Cell Biol 67(3): 606-622.
- Bloodgood, R. A. (2009). The Chlamydomonas flagellar membrane and its dynamic properties. The Chlamydomonas Sourcebook 2nd edition. 309-368.
- Bloodgood, R. A. (2012). The future of ciliary and flagellar membrane research. Mol Biol Cell 23(13): 2407-2411.
- Bloodgood, R. A. and May, G. S. (1982). Functional modification of the Chlamydomonas flagellar surface. J Cell Biol 93(1): 88-96.
- Dentler, W. L. (1980). Microtubule-membrane interactions in cilia. I. Isolation and characterization of ciliary membranes from Tetrahymena pyriformis. J Cell Biol 84(2): 364-380.
- Dentler, W. L. (1995). Isolation and fractionation of ciliary membranes from Tetrahymena. Methods Cell Biol 47: 397-400.
- *Dentler, W. (2013). A role for the membrane in regulating Chlamydomonas flagellar length. PLoS One 8(1): e53366.
- Harris, E. H. (1989). A comprehensive guide to biology and laboratory use. The Chlamydomonas Sourcebook. Academic Press.
- Huang, K., Diener, D. R., Mitchell, A., Pazour, G. J., Witman, G. B. and Rosenbaum, J. L. (2007). Function and dynamics of PKD2 in Chlamydomonas reinhardtii flagella. J Cell Biol 179(3): 501-514.
- Iomini, C., Li, L., Mo, W., Dutcher, S. K. and Piperno, G. (2006). Two flagellar genes, AGG2 and AGG3, mediate orientation to light in Chlamydomonas. Curr Biol 16(11): 1147-1153.
- Kalshoven, H., Musgrave, A. and Van den Ende, H. (1990). Mating receptor complex in the flagellar membrane of Chlamydomonas eugametos gametes. Sexual Plant Reproduction 3(2): 77-87.
- McLean, R.J., Laurendi, C. J. and Brown, R. M., Jr. (1974). The relationship of gamone to the mating reaction in Chlamydomonas moewusii. Proc Natl Acad Sci U S A 71(7): 2610-2613.
- Pazour, G. J. and Bloodgood, R. A. (2008). Targeting proteins to the ciliary membrane. Curr Top Dev Biol 85: 115-149.
- Pasquale, S. M. and Goodenough, U. W. (1987). Cyclic AMP functions as a primary sexual signal in gametes of Chlamydomonas reinhardtii. J Cell Biol 105(5): 2279-2292.
- Sager, R. and Granick, S. (1953). Nutritional studies with Chlamydomonas reinhardi. Ann N Y Acad Sci 56(5): 831-838.
- Snell, W. J. (1976). Mating in Chlamydomonas: a system for the study of specific cell adhesion. I. Ultrastructural and electrophoretic analyses of flagellar surface components involved in adhesion. J Cell Biol 68(1): 48-69.
- Wang, Q. and Snell, W. J. (2003). Flagellar adhesion between mating type plus and mating type minus gametes activates a flagellar protein-tyrosine kinase during fertilization in Chlamydomonas. J Biol Chem 278(35): 32936-32942.
- Witman, G. B., Carlson, K., Berliner, J. and Rosenbaum, J. L. (1972). Chlamydomonas flagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes. J Cell Biol 54(3): 507-539.
- Wood, C. R., Huang, K., Diener, D. R. and Rosenbaum, J. L. (2013). The cilium secretes bioactive ectosomes. Curr Biol 23(10): 906-911.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dentler, W. (2014). Ciliary and Flagellar Membrane Vesicle (Ectosome) Purification. Bio-protocol 4(12): e1156. DOI: 10.21769/BioProtoc.1156.
Category
Plant Science > Phycology > Cell analysis
Cell Biology > Cell movement > Cell motility
Cell Biology > Organelle isolation > Membrane
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.