- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Intracellular Glycogen Assays

Published: Vol 4, Iss 11, Jun 5, 2014 DOI: 10.21769/BioProtoc.1148 Views: 13801
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2013
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Glycogen, a soluble multi-branched glucose homopolysaccharide, is composed of chains of α-1,4-linked glucose residues interconnected by α-1,6-linked branches. The classical biosynthetic pathway involves phosphoglucomutase (Pgm), glucose-1-phosphate adenylyltransferase (GlgC or GlgCD), glycogen synthase (GlgA) and branching enzyme (GlgB). Phosphoglucomutase converts glucose-6-phosphate into glucose-1-phosphate, which serves as a substrate for ADP-glucose synthesis catalyzed by GlgC or GlgCD. Then, GlgA catalyzes the transfer of glucosyl units from ADP-glucose to the elongating chain of linear α-1,4-glucan. GlgB subsequently cleaves off portions of the glucan and links it to internal glucose molecules in existing chains via α-1,6 glycosidic bonds to form the glycogen structure. Glycogen breakdown is mediated by glycogen phosphorylase (GlgP) and debranching enzyme (GlgX), which catalyze the sequential phosphorolysis of α-1,4-glucosyl linkages in the glucan chain from the non-reducing ends and debranching of the limit dextrins generated by GlgP, respectively. An increasing number of studies have revealed the involvement of glycogen metabolism in a multitude of physiological functions in some prokaryotes beyond the function of synthesizing energy storage compounds. Lactobacillus acidophilus NCFM was the first probiotic lactic acid bacterium demonstrated to possess a functional glycogen biosynthesis pathway that is involved in its growth, bile tolerance and complex carbohydrate metabolism (Goh and Klaenhammer, 2013). The following qualitative (for detection of intracellular glycogen) and quantitative (for measurement of intracellular glycogen content) intracellular glycogen assay protocols for Lactobacillus acidophilus (L. acidophilus) were modified from previous works (Govons et al., 1969; Law et al., 1995; Parrou and Francois, 1997) and should be applicable to other lactic acid bacteria as well as most microorganisms.
Part I. Qualitative detection of intracellular glycogen with iodine-staining method
Materials and Reagents
- L. acidophilus strains, or desired bacterial strains
- MRS broth (Difco), or other liquid growth medium of choice (stored at 4 °C)
- Iodine solution (see Recipes)
- Solid semi-defined medium (SDM) with 2% (w/v) carbohydrate (see Recipes)
Equipment
- 37 °C incubator
- 37 °C anaerobic chamber incubator
- Pipettor
Procedure
- Inoculate L. acidophilus strains of interest in 5 ml of MRS broth and grow overnight at 37 °C under anaerobic or ambient atmospheric condition.
- Transfer and spot 5 to 8 µl of each overnight culture onto solid agar growth medium (we use solid SDM supplemented with 2% trehalose or glucose). Include positive and/or negative control strains, if possible.
Optional: The spotted plates can be prepared in multiple replicates to provide several iodine staining trials (see steps 4-6). - Allow the spotted cultures to absorb into the agar medium, and incubate the plate at 37 °C anaerobically for 36-48 h until colonies are visible.
- Carefully transfer 5 ml of freshly prepared iodine solution (0.01 M I2, 0.03 M KI) onto the agar plate with the grown colonies.
- Incubate the cells with the iodine solution at room temperature for 30 sec to 1 min.
- Immediately remove the iodine solution from the plate with a pipettor. Cells containing intracellular glycogen will stain brown; whereas cells that lack intracellular glycogen will appear yellow or colorless.
Optional: To enhance color development for capturing image of the stained colonies, the stained plates (after removal of iodine solution) can be incubated for an additional 10 min in the dark.
Note: The stain from iodine solution is not permanent and will gradually disappear after about 30 min to 1 h.
Representative data
- Example: Spotted cultures of L. acidophilus NCFM and glycogen metabolic mutants stained with iodine solution.
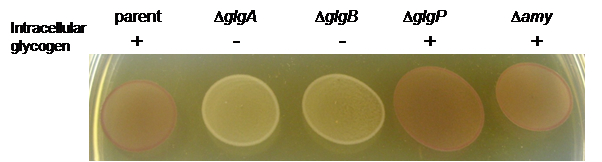
Figure 1. Iodine staining of L. acidophilus parent strain (NCK1909) and isogenic glycogen metabolic mutants grown on solid SDM containing 2% trehalose. Both deletion mutants of the glycogen synthesis pathway, ΔglgA and ΔglgB, appeared as yellow/colorless (-) indicative of glycogen-deficient phenotype. Like the parent strain, the ΔglgP and Δamy mutants of the glycogen degradation pathway were stained brown (+), indicating that their ability to synthesize intracellular glycogen was unaffected. glgA, glycogen synthase gene; glgB, branching enzyme gene; glgP, glycogen phosphorylase gene, and amy, putative α-amylase/debranching enzyme gene. Figure from Goh and Klaenhammer (2013).
Recipes
- Iodine solution
0.01 M I2
0.03 M KI
Prepare fresh prior to each experiment
Keep in dark - Semi-defined medium (SDM; modified from Reference 3)
Tween 80 (1 g/L)
Ammonium citrate (2 g/L)
Sodium acetate (5 g/L)
MgSO4.7H2O (0.1 g/L)
MnSO4 (0.05 g/L)
K2HPO4 (2 g/L)
Yeast nitrogen base (5 g/L)
Casitone (10 g/L)
Carbohydrate substrate, e.g. glucose or trehalose, 2% (or desired) final concentration
Note: For trehalose or any heat-labile sugars, prepare a 25-40% (w/v) stock solution, filter-sterilize, and add appropriate volume to autoclaved SDM to a final concentration of 2%. For carbohydrates with poor solubility e.g. raffinose, add 2% (w/v) of the sugar directly into SDM (sterile or non-sterile), stir to solubilize, and filter-sterilized through 0.45 µm filter.
For solid medium, add agar, 15 g/L
Sterilize by autoclaving or filter-sterilize with 0.45 µm filter (see above; for broth only)
Stored at 4 °C
Part II. Quantitative measurement of intracellular glycogen
Materials and Reagents
- L. acidophilus strains (or other bacterial strains)
- MRS broth (stored at 4 °C)
- Phosphate-buffered saline (PBS, pH 7.4) (e. g. Life Technologies, catalog number: 10010-023 )
- 0.25 M Na2CO3 solution (filter-sterilized with 0.45 µm filter)
- 1 M acetic acid
- 0.2 M sodium acetate (pH 5.2) (filter-sterilized with 0.45 µm filter)
- Sterile distilled water
- Amyloglucosidase (10 mg/ml ≈ 0.14 U/ µl) (Roche Diagnostics, catalog number: 10102857001 ) (stored at 4 °C)
- Glucose assay kit (Glucose assay reagent) (Sigma-Aldrich, catalog number: G3293 ) (stored at 4 °C)
- Glycogen (5-20 mg/ml solution) (e. g. Life Technologies, catalog number AM9510 ) (stored at -20 °C)
- SDM broth with 2% (w/v) carbohydrate substrate (see Recipes) (or liquid growth medium of choice)
Equipment
- 37 °C incubator
- Centrifuge (fits 15-ml and 50-ml tubes; 1,717 x g)
- Microcentrifuge (14,549 x g)
- Pipettor
- Disposable conical centrifuge tubes (15-ml and 50-ml)
- 1.5-ml screw-capped tubes (sterile) (e. g. Thermo Fisher Scientific, catalog numbers: 02-681-343 and 02-681-368 ) (autoclave tubes with caps on prior to use)
- 1.5-ml microcentrifuge tubes (sterile)
- Analytical balance
- Vortex
- Hot plate
- Large glass beaker (e. g. 1,000-ml pyrex glass beaker)
- Microcentrifuge tube floating rack (round, to fit into large glass beaker)
- Thermometer
- Incubator at 57 °C equipped with agitator (or hybridization oven)
- Spectrophotometer or microtiter plate reader (capable of measuring absorbance at 340 nm)
Procedure
- Cultivate L. acidophilus cells in SDM broth containing 2% (w/v) carbohydrate substrate (we supplemented with carbohydrate substrate that is known to induce glycogen biosynthesis) at 37 °C to desired growth phase and harvest by centrifugation (1,717 x g, 10 min, room temperature). Cell pellets can be processed right after centrifugation or stored at -80 °C.
Notes:- In general, for early log (OD~0.3 to 0.4), mid-log (OD~0.6), stationary (OD~1.0 to 1.6) and late stationary phase cultures (OD~ ≥ 2.0), we harvest aliquots of ca. 50 ml, 30 ml, 20 ml and 15 ml, respectively, in disposable conical centrifuge tubes, to yield ca. 0.04 to 0.06 g of cell wet weight.
- We prepare cultures and cell pellets representing at least two biological replicates in order to demonstrate reproducibility of the experimental results.
- In general, for early log (OD~0.3 to 0.4), mid-log (OD~0.6), stationary (OD~1.0 to 1.6) and late stationary phase cultures (OD~ ≥ 2.0), we harvest aliquots of ca. 50 ml, 30 ml, 20 ml and 15 ml, respectively, in disposable conical centrifuge tubes, to yield ca. 0.04 to 0.06 g of cell wet weight.
- Pre-label, pre-weigh (with cap on), and designate each 1.5-ml screw-capped tube for each sample.
- Resuspend each cell pellet in 1 ml of PBS and transfer to designated pre-weighed 1.5 ml screw-capped tube.
- Centrifuge (14,549 x g, 1 min, room temperature) and remove all supernatant with a pipettor.
- Repeat step 4 centrifugation for at least twice to remove all the supernatant in order to obtain an accurate cell wet weight.
- Weigh the tubes (with caps on) with the cell pellets. Subtract the previous weights of the empty tubes (with cap; from step 2 above) from the weight values to obtain net cell wet weight.
- Add 0.25 ml of 0.25 M Na2CO3 solution to the cell pellet. Resuspend by gentle vortexing.
Note: Do not resuspend the cell pellet with a pipettor since any amount of cells adhering to the pipet tip will change the net cell wet weight.
For controls: Include 2 additional tubes (no pre-weighing necessary). In each tube, add ~400 µg (e. g. 20 µl of 20 mg/ml) of glycogen to 0.25 ml of 0.25 M Na2CO3 solution. Follow steps 8-12. - Incubate the cell suspensions at 95 to 98 °C for 4 h (We place the tubes securely in a microcentrifuge tube floating rack and incubate in a large beaker of water heated on a hot plate.). Gently vortex to mix the cell suspension 1 to 2 times during the 4-h incubation.
- Add 0.15 ml of 1 M acetic acid and 0.6 ml of 0.2 M sodium acetate (pH 5.2) to the cell suspension to bring the pH to ~5.2 and the final volume to 1 ml.
- Add 10 µl of amyloglucosidase (0.14 U/µl) to the cell suspension (1.4 U/ml final concentration of amyloglucosidase).
For controls: Into the two control tubes containing 400 µg of glycogen (see step 7), add 10 µl of amyloglucosidase to one tube (labeled as positive control) and 10 µl of sterile distilled water to the remaining tube (labeled as negative control). - Incubate the tubes at 57 °C overnight with constant agitation (We seal the tubes with parafilm and incubate the tubes in a hybridization oven.).
- The next day, centrifuge the cell suspensions (5,000 x g, 3 min, room temperature) to pellet the cell debris.
- Measure the glucose released from the intracellular glycogen using a hexokinase/glucose-6-phosphate dehydrogenase-based glucose assay kit according to manufacturer’s instructions.
- The resulting glucose concentration (mg of glucose in 1 ml of the sample from step 9) is used to calculate the intracellular glycogen content per g of cell wet weight (expressed as mg of glucose per g of cell wet weight).
Representative data
- Example: Quantitative determination of intracellular glycogen contents in L. acidophilus parent strain (NCK1909) and isogenic glycogen metabolic mutants.
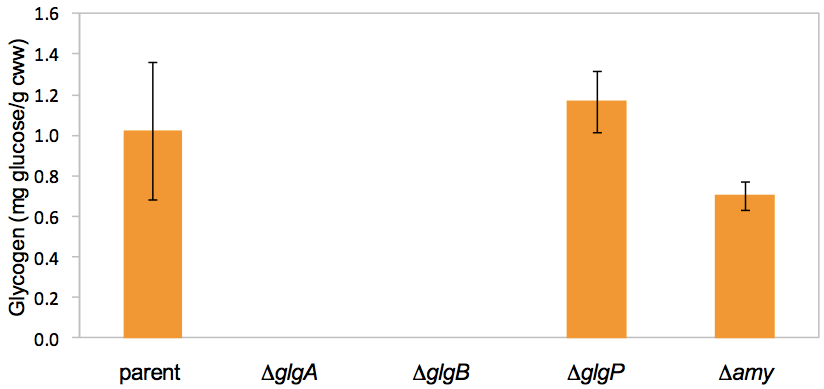
Figure 2. Intracellular glycogen contents of mid-log phase cells grown in SDM containing 2% trehalose. In this quantitative assay, glycogen was not detected from both ΔglgA and ΔglgB mutants. This confirms the results from the qualitative detection assay above using iodine-staining method (Figure 1). Data shown represent the mean ± standard deviation for two independent biological replicates. Figure adapted from Goh et al. (2003).
Recipes
- Semi-defined medium [(SDM; modified from Kimmel and Roberts (1998)]
Tween 80 (1 g/L)
Ammonium citrate (2 g/L)
Sodium acetate (5 g/L)
MgSO4.7H2O (0.1 g/L)
MnSO4 (0.05 g/L)
K2HPO4 (2 g/L)
Yeast nitrogen base (5 g/L)
Casitone (10 g/L)
Carbohydrate substrate, e.g. glucose or trehalose, 2% (or desired) final concentration
Note: For trehalose or any heat-labile sugars, prepare a 25-40% (w/v) stock solution, filter-sterilize, and add appropriate volume to autoclaved SDM to a final concentration of 2%. For carbohydrates with poor solubility e.g. raffinose, add 2% (w/v) of the sugar directly into SDM (sterile or non-sterile), stir to solubilize, and filter-sterilized through 0.45 µm filter.
For solid medium, add agar, 15 g/L
Sterilize by autoclaving or filter-sterilize with 0.45 µm filter (see above; for broth only)
Stored at 4 °C
Acknowledgments
The protocols were published in Goh and Klaenhammer (2013), and were developed based on modifications from the previous works of Govons et al. (1969), Kimmel and Roberts (1998), Law et al. (1995), and Parrou and Francois (1997). The funding sources for this work included Danisco/DuPont Nutrition and Health and the North Carolina Agricultural Foundation.
References
- Goh, Y. J. and Klaenhammer, T. R. (2013). A functional glycogen biosynthesis pathway in Lactobacillus acidophilus: expression and analysis of the glg operon. Mol Microbiol 89(6): 1187-1200.
- Govons, S., Vinopal, R., Ingraham, J. and Preiss, J. (1969). Isolation of mutants of Escherichia coli B altered in their ability to synthesize glycogen. J Bacteriol 97(2): 970-972.
- Kimmel, S. A. and Roberts, R. F. (1998). Development of a growth medium suitable for exopolysaccharide production by Lactobacillus delbrueckii ssp. bulgaricus RR. Int J Food Microbiol 40(1-2): 87-92.
- Law, J., Buist, G., Haandrikman, A., Kok, J., Venema, G. and Leenhouts, K. (1995). A system to generate chromosomal mutations in Lactococcus lactis which allows fast analysis of targeted genes. J Bacteriol 177(24): 7011-7018.
- Parrou, J. L. and Francois, J. (1997). A simplified procedure for a rapid and reliable assay of both glycogen and trehalose in whole yeast cells. Anal Biochem 248(1): 186-188.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Goh, Y. J. and Klaenhammer, T. R. (2014). Intracellular Glycogen Assays. Bio-protocol 4(11): e1148. DOI: 10.21769/BioProtoc.1148.
Category
Microbiology > Microbial metabolism > Carbohydrate
Biochemistry > Carbohydrate > Glycogen
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.